Mesonia Oceanica Sp. Nov., Isolated from Oceans During the Tara Oceans Expedition, with a Preference for Mesopelagic Waters
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chemical Structures of Some Examples of Earlier Characterized Antibiotic and Anticancer Specialized
Supplementary figure S1: Chemical structures of some examples of earlier characterized antibiotic and anticancer specialized metabolites: (A) salinilactam, (B) lactocillin, (C) streptochlorin, (D) abyssomicin C and (E) salinosporamide K. Figure S2. Heat map representing hierarchical classification of the SMGCs detected in all the metagenomes in the dataset. Table S1: The sampling locations of each of the sites in the dataset. Sample Sample Bio-project Site depth accession accession Samples Latitude Longitude Site description (m) number in SRA number in SRA AT0050m01B1-4C1 SRS598124 PRJNA193416 Atlantis II water column 50, 200, Water column AT0200m01C1-4D1 SRS598125 21°36'19.0" 38°12'09.0 700 and above the brine N "E (ATII 50, ATII 200, 1500 pool water layers AT0700m01C1-3D1 SRS598128 ATII 700, ATII 1500) AT1500m01B1-3C1 SRS598129 ATBRUCL SRS1029632 PRJNA193416 Atlantis II brine 21°36'19.0" 38°12'09.0 1996– Brine pool water ATBRLCL1-3 SRS1029579 (ATII UCL, ATII INF, N "E 2025 layers ATII LCL) ATBRINP SRS481323 PRJNA219363 ATIID-1a SRS1120041 PRJNA299097 ATIID-1b SRS1120130 ATIID-2 SRS1120133 2168 + Sea sediments Atlantis II - sediments 21°36'19.0" 38°12'09.0 ~3.5 core underlying ATII ATIID-3 SRS1120134 (ATII SDM) N "E length brine pool ATIID-4 SRS1120135 ATIID-5 SRS1120142 ATIID-6 SRS1120143 Discovery Deep brine DDBRINP SRS481325 PRJNA219363 21°17'11.0" 38°17'14.0 2026– Brine pool water N "E 2042 layers (DD INF, DD BR) DDBRINE DD-1 SRS1120158 PRJNA299097 DD-2 SRS1120203 DD-3 SRS1120205 Discovery Deep 2180 + Sea sediments sediments 21°17'11.0" -

Eelgrass Sediment Microbiome As a Nitrous Oxide Sink in Brackish Lake Akkeshi, Japan
Microbes Environ. Vol. 34, No. 1, 13-22, 2019 https://www.jstage.jst.go.jp/browse/jsme2 doi:10.1264/jsme2.ME18103 Eelgrass Sediment Microbiome as a Nitrous Oxide Sink in Brackish Lake Akkeshi, Japan TATSUNORI NAKAGAWA1*, YUKI TSUCHIYA1, SHINGO UEDA1, MANABU FUKUI2, and REIJI TAKAHASHI1 1College of Bioresource Sciences, Nihon University, 1866 Kameino, Fujisawa, 252–0880, Japan; and 2Institute of Low Temperature Science, Hokkaido University, Kita-19, Nishi-8, Kita-ku, Sapporo, 060–0819, Japan (Received July 16, 2018—Accepted October 22, 2018—Published online December 1, 2018) Nitrous oxide (N2O) is a powerful greenhouse gas; however, limited information is currently available on the microbiomes involved in its sink and source in seagrass meadow sediments. Using laboratory incubations, a quantitative PCR (qPCR) analysis of N2O reductase (nosZ) and ammonia monooxygenase subunit A (amoA) genes, and a metagenome analysis based on the nosZ gene, we investigated the abundance of N2O-reducing microorganisms and ammonia-oxidizing prokaryotes as well as the community compositions of N2O-reducing microorganisms in in situ and cultivated sediments in the non-eelgrass and eelgrass zones of Lake Akkeshi, Japan. Laboratory incubations showed that N2O was reduced by eelgrass sediments and emitted by non-eelgrass sediments. qPCR analyses revealed that the abundance of nosZ gene clade II in both sediments before and after the incubation as higher in the eelgrass zone than in the non-eelgrass zone. In contrast, the abundance of ammonia-oxidizing archaeal amoA genes increased after incubations in the non-eelgrass zone only. Metagenome analyses of nosZ genes revealed that the lineages Dechloromonas-Magnetospirillum-Thiocapsa and Bacteroidetes (Flavobacteriia) within nosZ gene clade II were the main populations in the N2O-reducing microbiome in the in situ sediments of eelgrass zones. -

Genome Analysis of Flaviramulus Ichthyoenteri Th78t in the Family
Zhang et al. BMC Genomics (2015) 16:38 DOI 10.1186/s12864-015-1275-0 RESEARCH ARTICLE Open Access Genome analysis of Flaviramulus ichthyoenteri Th78T in the family Flavobacteriaceae: insights into its quorum quenching property and potential roles in fish intestine Yunhui Zhang1, Jiwen Liu1, Kaihao Tang1, Min Yu1, Tom Coenye2 and Xiao-Hua Zhang1* Abstract Background: Intestinal microbes play significant roles in fish and can be possibly used as probiotics in aquaculture. In our previous study, Flaviramulus ichthyoenteri Th78T, a novel species in the family Flavobacteriaceae, was isolated from fish intestine and showed strong quorum quenching (QQ) ability. To identify the QQ enzymes in Th78T and explore the potential roles of Th78T in fish intestine, we sequenced the genome of Th78T and performed extensive genomic analysis. Results: An N-acyl homoserine lactonase FiaL belonging to the metallo-β-lactamase superfamily was identified and the QQ activity of heterologously expressed FiaL was confirmed in vitro. FiaL has relatively little similarity to the known lactonases (25.2 ~ 27.9% identity in amino acid sequence). Various digestive enzymes including alginate lyases and lipases can be produced by Th78T, and enzymes essential for production of B vitamins such as biotin, riboflavin and folate are predicted. Genes encoding sialic acid lyases, sialidases, sulfatases and fucosidases, which contribute to utilization of mucus, are present in the genome. In addition, genes related to response to different stresses and gliding motility were also identified. Comparative genome analysis shows that Th78T has more specific genes involved in carbohydrate transport and metabolism compared to other two isolates in Flavobacteriaceae, both isolated from sediments. -

High Quality Permanent Draft Genome Sequence of Chryseobacterium Bovis DSM 19482T, Isolated from Raw Cow Milk
Lawrence Berkeley National Laboratory Recent Work Title High quality permanent draft genome sequence of Chryseobacterium bovis DSM 19482T, isolated from raw cow milk. Permalink https://escholarship.org/uc/item/4b48v7v8 Journal Standards in genomic sciences, 12(1) ISSN 1944-3277 Authors Laviad-Shitrit, Sivan Göker, Markus Huntemann, Marcel et al. Publication Date 2017 DOI 10.1186/s40793-017-0242-6 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Laviad-Shitrit et al. Standards in Genomic Sciences (2017) 12:31 DOI 10.1186/s40793-017-0242-6 SHORT GENOME REPORT Open Access High quality permanent draft genome sequence of Chryseobacterium bovis DSM 19482T, isolated from raw cow milk Sivan Laviad-Shitrit1, Markus Göker2, Marcel Huntemann3, Alicia Clum3, Manoj Pillay3, Krishnaveni Palaniappan3, Neha Varghese3, Natalia Mikhailova3, Dimitrios Stamatis3, T. B. K. Reddy3, Chris Daum3, Nicole Shapiro3, Victor Markowitz3, Natalia Ivanova3, Tanja Woyke3, Hans-Peter Klenk4, Nikos C. Kyrpides3 and Malka Halpern1,5* Abstract Chryseobacterium bovis DSM 19482T (Hantsis-Zacharov et al., Int J Syst Evol Microbiol 58:1024-1028, 2008) is a Gram-negative, rod shaped, non-motile, facultative anaerobe, chemoorganotroph bacterium. C. bovis is a member of the Flavobacteriaceae, a family within the phylum Bacteroidetes. It was isolated when psychrotolerant bacterial communities in raw milk and their proteolytic and lipolytic traits were studied. Here we describe the features of this organism, together with the draft genome sequence and annotation. The DNA G + C content is 38.19%. The chromosome length is 3,346,045 bp. It encodes 3236 proteins and 105 RNA genes. The C. bovis genome is part of the Genomic Encyclopedia of Type Strains, Phase I: the one thousand microbial genomes study. -

Genome Analysis and Classification of Novel Species Flavobacterium Gabrieli
NOTICE: The copyright law of the United States (Title 17, United States Code) governs the making of reproductions of copyrighted material. One specified condition is that the reproduction is not to be "used for any purpose other than private study, scholarship, or research." If a user makes a request for, or later uses a reproduction for purposes in excess of "fair use," that user may be liable for copyright infringement. RESTRICTIONS: This student work may be read, quoted from, cited, for purposes of research. It may not be published in full except by permission of the author. 1 Kirsten Fischer Introduction Microbial Systematics and Taxonomy The diversity of bacteria is truly immense and the discovery of new species and higher taxonomic groups happens quite frequently, as evidenced by the ever expanding tree of life (Hug et al., 2016). The classification of prokaryotes, bacteria especially, is formally regulated by the International Committee on the Systematics of Prokaryotes and has experienced rapid change over the last fifty years. However, some feel that these rules could be even stricter for proper organization of taxonomy (Tindall et al., 2010). Problems occur with the integration of newer methodologies, which creates some challenges for the researcher attempting to publish a novel species. For example, some DNA sequences that are deposited in databases are not accurate (Clarridge, 2004). Taxonomy is an artificial system that works based on the intuition of scientists rather than strict, specific standards (Konstantinidis & Tiedje, 2005). Tindall advocates that a strain shown to be a novel taxon should be characterized “as comprehensively as possible” and abide by the framework established in the Bacteriological Code (2010). -
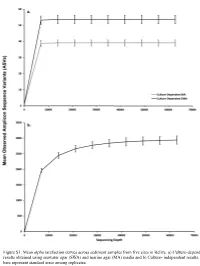
Figure S1. Mean Alpha Rarefaction Curves Across Sediment Samples from Five Sites in Belize
Figure S1. Mean alpha rarefaction curves across sediment samples from five sites in Belize. a) Culture-dependent results obtained using seawater agar (SWA) and marine agar (MA) media and b) Culture- independent results. Error bars represent standard error among replicates. a. 200 150 100 Faith’s PD Faith’s 50 0 Culturedependent MA Culturedependent SWA Cultureindependent Method b. 0.8 0.6 Pielou’s Evenness 0.4 Culturedependent MA Culturedependent SWA Cultureindependent Method Figure S2. Alpha diversity boxplots of marine sediment microbial communities from Carrie Bow Cay, Belize in culture-dependent and culture-independent samples determined using a) Faith’s Phylogenetic Diversity Index and b) Pielou’s Evenness. Culture-dependent methods include the use of marine agar medium (MA) and seawater agar medium (SWA. Data points are overlayed on the boxplot to show variation. a. 100% Proteobacteria Nitrospinae Bacteroidetes Fibrobacteres Planctomycetes Entotheonellaeota 90% Cyanobacteria Marinimicrobia (SAR406 clade) Firmicutes Deinococcus-Thermus Chloroflexi Margulisbacteria [A] Thaumarchaeota Unassigned 80% Acidobacteria Archaea UA Verrucomicrobia Acetothermia Actinobacteria Elusimicrobia 70% [A] Nanoarchaeaeota Tenericutes Kiritimatiellaeota TA06 Latescibacteria WS2 60% Spirochaetes Dependentiae Patescibacteria LCP-89 Gemmatimonadetes FCPU426 Omnitrophicaeota WPS-2 50% Fusobacteria CK-2C2-2 Bacteria UA Armatimonadetes [A] Euryarchaeota [A] Altiarchaeota Relative Percent 40% Calditrichaeota Poribacteria Lentisphaerae Cloacimonetes [A] Crenarchaeota -

Muricauda Ruestringensis Type Strain (B1T)
Standards in Genomic Sciences (2012) 6:185-193 DOI:10.4056/sigs.2786069 Complete genome sequence of the facultatively anaerobic, appendaged bacterium Muricauda T ruestringensis type strain (B1 ) Marcel Huntemann1, Hazuki Teshima1,2, Alla Lapidus1, Matt Nolan1, Susan Lucas1, Nancy Hammon1, Shweta Deshpande1, Jan-Fang Cheng1, Roxanne Tapia1,2, Lynne A. Goodwin1,2, Sam Pitluck1, Konstantinos Liolios1, Ioanna Pagani1, Natalia Ivanova1, Konstantinos Mavromatis1, Natalia Mikhailova1, Amrita Pati1, Amy Chen3, Krishna Palaniappan3, Miriam Land1,4 Loren Hauser1,4, Chongle Pan1,4, Evelyne-Marie Brambilla5, Manfred Rohde6, Stefan Spring5, Markus Göker5, John C. Detter1,2, James Bristow1, Jonathan A. Eisen1,7, Victor Markowitz3, Philip Hugenholtz1,8, Nikos C. Kyrpides1, Hans-Peter Klenk5*, and Tanja Woyke1 1 DOE Joint Genome Institute, Walnut Creek, California, USA 2 Los Alamos National Laboratory, Bioscience Division, Los Alamos, New Mexico, USA 3 Biological Data Management and Technology Center, Lawrence Berkeley National Laboratory, Berkeley, California, USA 4 Oak Ridge National Laboratory, Oak Ridge, Tennessee, USA 5 Leibniz Institute DSMZ - German Collection of Microorganisms and Cell Cultures, Braunschweig, Germany 6 HZI – Helmholtz Centre for Infection Research, Braunschweig, Germany 7 University of California Davis Genome Center, Davis, California, USA 8 Australian Centre for Ecogenomics, School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Australia *Corresponding author: Hans-Peter Klenk ([email protected]) Keywords: facultatively anaerobic, non-motile, Gram-negative, mesophilic, marine, chemo- heterotrophic, Flavobacteriaceae, GEBA Muricauda ruestringensis Bruns et al. 2001 is the type species of the genus Muricauda, which belongs to the family Flavobacteriaceae in the phylum Bacteroidetes. The species is of inter- est because of its isolated position in the genomically unexplored genus Muricauda, which is located in a part of the tree of life containing not many organisms with sequenced genomes. -

Table S5. the Information of the Bacteria Annotated in the Soil Community at Species Level
Table S5. The information of the bacteria annotated in the soil community at species level No. Phylum Class Order Family Genus Species The number of contigs Abundance(%) 1 Firmicutes Bacilli Bacillales Bacillaceae Bacillus Bacillus cereus 1749 5.145782459 2 Bacteroidetes Cytophagia Cytophagales Hymenobacteraceae Hymenobacter Hymenobacter sedentarius 1538 4.52499338 3 Gemmatimonadetes Gemmatimonadetes Gemmatimonadales Gemmatimonadaceae Gemmatirosa Gemmatirosa kalamazoonesis 1020 3.000970902 4 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas indica 797 2.344876284 5 Firmicutes Bacilli Lactobacillales Streptococcaceae Lactococcus Lactococcus piscium 542 1.594633558 6 Actinobacteria Thermoleophilia Solirubrobacterales Conexibacteraceae Conexibacter Conexibacter woesei 471 1.385742446 7 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas taxi 430 1.265115184 8 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas wittichii 388 1.141545794 9 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas sp. FARSPH 298 0.876754244 10 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sorangium cellulosum 260 0.764953367 11 Proteobacteria Deltaproteobacteria Myxococcales Polyangiaceae Sorangium Sphingomonas sp. Cra20 260 0.764953367 12 Proteobacteria Alphaproteobacteria Sphingomonadales Sphingomonadaceae Sphingomonas Sphingomonas panacis 252 0.741416341 -

Revealing the Full Biosphere Structure and Versatile Metabolic Functions In
Chen et al. Genome Biology (2021) 22:207 https://doi.org/10.1186/s13059-021-02408-w RESEARCH Open Access Revealing the full biosphere structure and versatile metabolic functions in the deepest ocean sediment of the Challenger Deep Ping Chen1†, Hui Zhou1,2†, Yanyan Huang3,4†, Zhe Xie5†, Mengjie Zhang1,2†, Yuli Wei5, Jia Li1,2, Yuewei Ma3, Min Luo5, Wenmian Ding3, Junwei Cao5, Tao Jiang1,2, Peng Nan3*, Jiasong Fang5* and Xuan Li1,2* * Correspondence: nanpeng@fudan. edu.cn; [email protected]; lixuan@ Abstract sippe.ac.cn †Ping Chen, Hui Zhou, Yanyan Background: The full biosphere structure and functional exploration of the microbial Huang, Zhe Xie and Mengjie Zhang communities of the Challenger Deep of the Mariana Trench, the deepest known contributed equally to this work. hadal zone on Earth, lag far behind that of other marine realms. 3Ministry of Education Key Laboratory for Biodiversity Science Results: We adopt a deep metagenomics approach to investigate the microbiome and Ecological Engineering, School in the sediment of Challenger Deep, Mariana Trench. We construct 178 of Life Sciences, Fudan University, Shanghai, China metagenome-assembled genomes (MAGs) representing 26 phyla, 16 of which are 5Shanghai Engineering Research reported from hadal sediment for the first time. Based on the MAGs, we find the Center of Hadal Science and microbial community functions are marked by enrichment and prevalence of Technology, College of Marine Sciences, Shanghai Ocean mixotrophy and facultative anaerobic metabolism. The microeukaryotic community is University, Shanghai, China found to be dominated by six fungal groups that are characterized for the first time 1CAS-Key Laboratory of Synthetic in hadal sediment to possess the assimilatory and dissimilatory nitrate/sulfate Biology, CAS Center for Excellence in Molecular Plant Sciences, Institute reduction, and hydrogen sulfide oxidation pathways. -

Habitat and Taxon As Driving Forces of Carbohydrate
Habitat and taxon as driving forces of carbohydrate catabolism in marine heterotrophic bacteria: example of the model algae-associated bacterium Zobellia galactanivorans Dsij T Tristan Barbeyron, François Thomas, Valérie Barbe, Hanno Teeling, Chantal Schenowitz, Carole Dossat, Alexander Goesmann, Catherine Leblanc, Frank Oliver Glöckner, Mirjam Czjzek, et al. To cite this version: Tristan Barbeyron, François Thomas, Valérie Barbe, Hanno Teeling, Chantal Schenowitz, et al.. Habi- tat and taxon as driving forces of carbohydrate catabolism in marine heterotrophic bacteria: example of the model algae-associated bacterium Zobellia galactanivorans Dsij T. Environmental Microbiol- ogy, Society for Applied Microbiology and Wiley-Blackwell, 2016, Ecology and Physiology of Marine Microbes, 18 (12), pp.4610-4627. 10.1111/1462-2920.13584. hal-02137896 HAL Id: hal-02137896 https://hal.archives-ouvertes.fr/hal-02137896 Submitted on 23 May 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 Environmental Microbiology ‐ Research Article 2 3 Habitat and taxon -

Salegentibacter Agarivorans Sp. Nov., a Novel Marine Bacterium of the Family Flavobacteriaceae Isolated from the Sponge Artemisina Sp
International Journal of Systematic and Evolutionary Microbiology (2006), 56, 883–887 DOI 10.1099/ijs.0.64167-0 Salegentibacter agarivorans sp. nov., a novel marine bacterium of the family Flavobacteriaceae isolated from the sponge Artemisina sp. Olga I. Nedashkovskaya,1 Seung Bum Kim,2 Marc Vancanneyt,3 Dong Sung Shin,2 Anatoly M. Lysenko,4 Lyudmila S. Shevchenko,1 Vladimir B. Krasokhin,1 Valery V. Mikhailov,1 Jean Swings3 and Kyung Sook Bae5 Correspondence 1Pacific Institute of Bioorganic Chemistry of the Far-Eastern Branch of the Russian Academy Olga I. Nedashkovskaya of Sciences, Pr. 100 Let Vladivostoku 159, 690022, Vladivostok, Russia [email protected] 2Department of Microbiology, School of Bioscience and Biotechnology, Chungnam National University, 220 Gung-dong, Yusong, Daejon 305-764, Republic of Korea 3BCCM/LMG Bacteria Collection, Laboratory of Microbiology, Ghent University, Ledeganckstraat 35, B-9000 Ghent, Belgium 4Institute of Microbiology of the Russian Academy of Sciences, Pr. 60 Let October 7/2, Moscow, 117811, Russia 5Korea Research Institute of Bioscience and Biotechnology, 52 Oun-Dong, Yusong, Daejon 305-333, Republic of Korea A sponge-associated strain, KMM 7019T, was investigated in a polyphasic taxonomic study. The bacterium was strictly aerobic, heterotrophic, Gram-negative, yellow-pigmented, motile by gliding and oxidase-, catalase-, b-galactosidase- and alkaline phosphatase-positive. A phylogenetic analysis based on 16S rRNA gene sequences revealed that strain KMM 7019T is closely related to members of the genus Salegentibacter, namely Salegentibacter holothuriorum, Salegentibacter mishustinae and Salegentibacter salegens (97?7–98 % sequence similarities). The DNA–DNA relatedness between the strain studied and Salegentibacter species ranged from 27 to 31 %, clearly demonstrating that KMM 7019T belongs to a novel species of the genus Salegentibacter, for which the name Salegentibacter agarivorans sp. -

Flavobacterial Response to Organic Pollution
Vol. 51: 31–43, 2008 AQUATIC MICROBIAL ECOLOGY Published April 24 doi: 10.3354/ame01174 Aquat Microb Ecol Flavobacterial response to organic pollution Andrew Bissett1, 2, 3,*, John P. Bowman2, Chris M. Burke1 1School of Aquaculture, Tasmanian Aquaculture and Fisheries Institute, University of Tasmania and Aquafin CRC, Launceston, Tasmania 7250, Australia 2School of Agricultural Science, University of Tasmania, Hobart, Tasmania 7001, Australia 3Present address: Max Planck Institute for Marine Microbiology, Celsiusstrasse 1, 28359 Bremen, Germany ABSTRACT: Bacteria of the Cytophaga-Flavobacterium-Bacteroides (CFB) group (phylum Bac- teroidetes), in particular members of the class Flavobacteria, are among the most prominent hetero- trophic organisms in marine pelagic systems. They have also previously been found to be important in the initial biopolymer degradation of sedimentary organic matter. The Flavobacteria community was analysed in inshore, marine sediments subject to regular inputs of highly labile organic carbon in order to understand the importance of this group in carbon degradation. We used denaturing gra- dient gel electrophoresis and real-time PCR in a statistically robust manner, over 2 consecutive years, to demonstrate that the number of Flavobacteria in the sediment increased and community composi- tion shifted with organic loading. Further community shifts occurred after cessation of organic load- ing, and population numbers also decreased. Flavobacteria appear to be important in the initial responses of the sediment microbial community to organic loading, regardless of sediment type, but flavobacterial composition was not predictable. The highly dynamic nature and large diversity (func- tional redundancy) of the Flavobacteria in these sediments may contribute to this unpredictable response. KEY WORDS: Flavobacteria .