Functional Characterization of the Interaction Between Bex2 and Torsina, a Protein Involved in Early-Onset Torsion Dystonia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Role of Structural Disorder in Cell Cycle Regulation, Related Clinical Proteomics, 5 Disease Development and Drug Targeting
Review The role of structural disorder in cell cycle regulation, related clinical proteomics, 5 disease development and drug targeting Expert Rev. Proteomics 12(3), 000–000 (2015) 10 1 AQ2 Agnes Tantos , Understanding the molecular mechanisms of the regulation of cell cycle is a central issue in Lajos Kalmar2 and molecular cell biology, due to its fundamental role in the existence of cells. The regulatory Peter Tompa*1,2 circuits that make decisions on when a cell should divide are very complex and particularly subtly balanced in eukaryotes, in which the harmony of many different cells in an organism is 1 Institute of Enzymology, Research essential for life. Several hundred proteins are involved in these processes, and a great deal of 15 Centre for Natural Sciences of the Hungarian Academy of Sciences, studies attests that most of them have functionally relevant intrinsic structural disorder. Budapest, Hungary Structural disorder imparts many functional advantages on these proteins, and we discuss it 2 VIB Department of Structural Biology, in detail that it is involved in all key steps from signaling through the cell membrane to Vrije Universiteit Brussel, Brussels, Belgium regulating transcription of proteins that execute timely responses to an ever-changing *Author for correspondence: environment. 20 [email protected] KEYWORDS: cancer . cell-cycle . checkpoint . post-translational modification . protein disorder . signal transduction 25 of proteins are able to fulfill important func- Cell cycle: the cornerstone of tions without possessing a stable three- multicellular life dimensional structure [3,4]. These proteins, Every postembryonic eukaryotic cell goes termed intrinsically disordered proteins or through the distinct phases of cell cycle, regions (IDPs/IDRs), participate in many reg- G1, S, G2 and M. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -

Haploinsufficiency of Cardiac Myosin Binding Protein-C in the Development of Hypertrophic Cardiomyopathy
Loyola University Chicago Loyola eCommons Dissertations Theses and Dissertations 2014 Haploinsufficiency of Cardiac Myosin Binding Protein-C in the Development of Hypertrophic Cardiomyopathy David Barefield Loyola University Chicago Follow this and additional works at: https://ecommons.luc.edu/luc_diss Part of the Physiology Commons Recommended Citation Barefield, David, "Haploinsufficiency of Cardiac Myosin Binding Protein-C in the Development of Hypertrophic Cardiomyopathy" (2014). Dissertations. 1249. https://ecommons.luc.edu/luc_diss/1249 This Dissertation is brought to you for free and open access by the Theses and Dissertations at Loyola eCommons. It has been accepted for inclusion in Dissertations by an authorized administrator of Loyola eCommons. For more information, please contact [email protected]. This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 3.0 License. Copyright © 2014 David Barefield LOYOLA UNIVERSITY CHICAGO HAPLOINSUFFICIENCY OF CARDIAC MYOSIN BINDING PROTEIN-C IN THE DEVELOPMENT OF HYPERTROPHIC CARDIOMYOPATHY A DISSERTATION SUBMITTED TO THE FACULTY OF THE GRADUATE SCHOOL IN CANDIDACY FOR THE DEGREE OF DOCTOR OF PHILOSOPHY PROGRAM IN CELL AND MOLECULAR PHYSIOLOGY BY DAVID YEOMANS BAREFIELD CHICAGO, IL AUGUST 2014 Copyright by David Yeomans Barefield, 2014 All Rights Reserved. ii ACKNOWLEDGEMENTS The completion of this work would not have been possible without the support of excellent mentors, colleagues, friends, and family. I give tremendous thanks to my mentor, Dr. Sakthivel Sadayappan, who has facilitated my growth as a scientist and as a human being over the past five years. I would like to thank my dissertation committee: Drs. Pieter de Tombe, Kenneth Byron, Leanne Cribbs, Kyle Henderson, and Christine Seidman for their erudite guidance of my project and my development as a scientist. -

Locating Gene Conversions on the X-Chromosome
Sexy Gene Conversions: Locating Gene Conversions on the X-Chromosome Mark J. Lawson1, Liqing Zhang1;2∗ Department of Computer Science, Virginia Tech 2Program in Genetics, Bioinformatics, and Computational Biology ∗To whom correspondence should be addressed; E-mail: [email protected] April 3, 2009 Abstract Gene conversion can have a profound impact on both the short-term and long-term evolution of genes and genomes. Here we examined the gene families that are located on the X-chromosomes of human, chimp, mouse, and rat for evidence of gene conversion. We identified seven gene families (WD repeat protein family, Ferritin Heavy Chain family, RAS-related Protein RAB-40 family, Diphosphoinositol polyphosphate phosphohydrolase family, Transcription Elongation Factor A family, LDOC1 Related family, Zinc Finger Protein ZIC, and GLI family) that show evidence of gene conversion. Through phylogenetic analyses and synteny evidence, we show that gene conversion has played an important role in the evolution of these gene families and that gene conversion has occured independently in both primates and rodents. Comparing the results with those of two gene conversion prediction programs (GENECONV and Partimatrix), we found that both GENECONV and Partimatrix have very high false negative rates (i.e. failed to predict gene conversions), leading to many undetected gene conversions. The combination of phylogenetic analyses with physical synteny evidence exhibits high power in the detection of gene conversions. 1 1 Introduction Gene conversions are the exchange of genetic information between two genes, initiated by a double-strand break in one gene (acceptor) followed by the repair of this gene through the copying of the sequence of a similar gene (donor). -

I FOUR JOINTED BOX ONE, a NOVEL PRO-ANGIOGENIC PROTEIN IN
FOUR JOINTED BOX ONE, A NOVEL PRO-ANGIOGENIC PROTEIN IN COLORECTAL CARCINOMA. BY Nicole Theresa Al-Greene Dissertation Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY In Cell and Developmental Biology. December, 2013 Nashville Tennessee Approved: R. Daniel Beauchamp Susan Wente James Goldenring Albert Reynolds i DEDICATION To my parents, Karen and John, who have helped me in every way possible, every single day. ii ACKNOWLEDGMENTS. Funding for this work was supported by grants DK052334, CA069457, The GI Cancer SPORE, GM088822, the VICC, the Clinical and Translational Science Award (NCRR/NIH UL1RR024975), the DDRC (P30DK058404), and the Cooperative Human Tissue Network (UO1CA094664) and U01CA094664. I was lucky enough to be allowed to perform research as an undergraduate in the lab of Ken Belanger. I will be forever grateful for that opportunity that sparked my love of research. Equally important was my time as a technician in Len Zon’s lab where I confirmed the fact that I needed to go graduate school and earn my degree. During my time at Vanderbilt I have been helped by so many individuals, and the collaborative nature of everyone I have met with is truly an amazing aspect of the research community here. I am especially thankful for all the technical help and insightful conversations I have had with Natasha Deane, Anna Means, Claudia Andl, Tanner Freeman, Connie Weaver, Keeli Lewis, Jalal Hamaamen, Jenny Zi, John Neff, Christian Kis, Andries Zjistra, Trennis Palmer, Joseph Roland, and Lynn LaPierre. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -

Atlas Journal
Atlas of Genetics and Cytogenetics in Oncology and Haematology Home Genes Leukemias Solid Tumours Cancer-Prone Deep Insight Portal Teaching X Y 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 NA Atlas Journal Atlas Journal versus Atlas Database: the accumulation of the issues of the Journal constitutes the body of the Database/Text-Book. TABLE OF CONTENTS Volume 12, Number 4, Jul-Aug 2008 Previous Issue / Next Issue Genes AKR1C3 (aldo-keto reductase family 1, member C3 (3-alpha hydroxysteroid dehydrogenase, type II)) (10p15.1). Hsueh Kung Lin. Atlas Genet Cytogenet Oncol Haematol 2008; Vol (12): 498-502. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/AKR1C3ID612ch10p15.html CASP1 (caspase 1, apoptosis-related cysteine peptidase (interleukin 1, beta, convertase)) (11q22.3). Yatender Kumar, Vegesna Radha, Ghanshyam Swarup. Atlas Genet Cytogenet Oncol Haematol 2008; Vol (12): 503-518. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/CASP1ID145ch11q22.html GCNT3 (glucosaminyl (N-acetyl) transferase 3, mucin type) (15q21.3). Prakash Radhakrishnan, Pi-Wan Cheng. Atlas Genet Cytogenet Oncol Haematol 2008; Vol (12): 519-524. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/GCNT3ID44105ch15q21.html HYAL2 (Hyaluronoglucosaminidase 2) (3p21.3). Lillian SN Chow, Kwok-Wai Lo. Atlas Genet Cytogenet Oncol Haematol 2008; Vol (12): 525-529. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/HYAL2ID40904ch3p21.html LMO2 (LIM domain only 2 (rhombotin-like 1)) (11p13) - updated. Pieter Van Vlierberghe, Jean Loup Huret. Atlas Genet Cytogenet Oncol Haematol 2008; Vol (12): 530-535. [Full Text] [PDF] URL : http://atlasgeneticsoncology.org/Genes/RBTN2ID34.html PEBP1 (phosphatidylethanolamine binding protein 1) (12q24.23). -

Protein Bex1 and Liver Regeneration After Tissue Damage
Università di Pisa Facoltà di Scienze Matematiche, Fisiche e Naturali Laurea Magistrale in Biologia Applicata alla Biomedicina Curriculum Fisiopatologico Protein Bex1 and Liver Regeneration after Tissue Damage Relatore: Candidato: Prof. Aldo Paolicchi Andrea Giannuzzo Relatore esterno: Prof.ssa Hanne Cathrine Bisgaard Anno Accademico 2010 - 2011 Index ABSTRACT ....................................................................................... pag. 4 1 INTRODUCTION ........................................................................... pag. 6 1.1 Brain Expressed X - Linked Gene Family ................................... pag. 6 1.2 Discovery and Localization ....................................................... pag. 7 1.3 Bex1 and p75NTR ...................................................................... pag. 11 1.4 Bex1 during Skeletal Muscle Regeneration ................................ pag. 15 1.5 Bex1 during Axonal Regeneration ............................................. pag. 18 1.6 Bex1 as Tumor Suppressor ........................................................ pag. 21 1.7 Architecture of the Liver and Regeneration ............................... pag. 24 1.7.1 Architecture .................................................................... pag. 24 1.7.2 Regeneration .................................................................... pag. 25 2 MATERIALS AND METHODS ......................................................... pag. 30 2.1 RNA Isolation ........................................................................... -
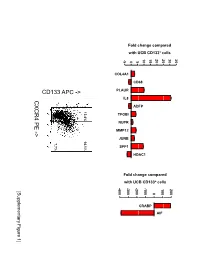
Supplementary Data
Fold change compared with UCB CD133+ cells 10 15 20 25 30 35 -5 0 5 COL4A1 CD68 PLAUR IL8 CXCR4 PE ADFP 13.4% 14.1% TFGBI PE NUPR - MMP12 > JUNB 7.7% SPP1 HDAC1 Fold change compared with UCB CD133+ cells -400 -300 -200 -100 100 200 [Supplementary Figure1] 0 CRABP AIF Rutella S et al. Supplementary Materials and Methods Real-time PCR Real-time quantitative PCR (qPCR) was performed on mRNA samples isolated from either endometrial cancer-derived CD133+ cells or umbilical cord blood CD133+ cells with the RNeasy plus Mini Kit (QIAGEN, Hilden, Germany), as already detailed. Quantitative PCR for a selected group of genes was performed on the iCycler iQ system (Bio-Rad, Hercules, CA). Primer sets were designed using the Beacon Design Software (Version 3) and the sequences available in the GeneBankTM data base. The specific oligonucleotide primer sequences are detailed in Supplementary Table 8. Complementary DNA (cDNA) was prepared starting from 1μg of total RNA using the iScrypt cDNA Synthesis Kit, which contains RNase H + MMLV reverse transcriptase random primers and 5x reaction mixture (Bio-Rad), according to the manufacturer’s instructions. Reactions were conducted in the PTC-0200 DNA Engine (MJ RESEARCH). Amplification was carried out in a total volume of 25μl containing 0.3μM of each specific primer, 12,5μl 2X SYBR Green Master Mix (100mM KCl, 40mM Tris-HCl, pH 8.4, 0.4mM of each dNTP, iTaq DNA polymerase, 6mM MgCl2, SYBR Green I, 20nM fluorescein, stabilizers; Bio-Rad) and 2μl of diluted cDNA. The PCR reactions were cycled starting with a 3-minute template denaturation step at 95°C followed by 40 cycles of 15 seconds at 95°C and 1 minute at 60°C. -

Stringdb Package Vignette
STRINGdb Package Vignette Andrea Franceschini 15 March 2015 1 INTRODUCTION STRING (https://www.string-db.org) is a database of known and predicted protein-protein interac- tions. The interactions include direct (physical) and indirect (functional) associations. The database contains information from numerous sources, including experimental repositories, computational pre- diction methods and public text collections. Each interaction is associated with a combined confidence score that integrates the various evidences. We currently cover over 24 milions proteins from 5090 organisms. As you will learn in this guide, the STRING database can be usefull to add meaning to list of genes (e.g. the best hits coming out from a screen or the most differentially expressed genes coming out from a Microarray/RNAseq experiment.) We provide the STRINGdb R package in order to facilitate our users in accessing the STRING database from R. In this guide we explain, with examples, most of the package's features and function- alities. In the STRINGdb R package we use the new ReferenceClasses of R (search for "ReferenceClasses" in the R documentation.). Besides we make use of the iGraph package (http://igraph.sourceforge.net) as a data structure to represent our protein-protein interaction network. To begin, you should first know the NCBI taxonomy identifiers of the organism on which you have performed the experiment (e.g. 9606 for Human, 10090 for mouse). If you don't know that, you can search the NCBI Taxonomy (http://www.ncbi.nlm.nih.gov/taxonomy) or start looking at our species table (that you can also use to verify that your organism is represented in the STRING database). -

5779 Protein Identified, 4414 with 2 Or More Peptides, FDR 0.01
Proteomic anlysis of human PHEO a PGL (2 x PHEO, 1 x PGL and control adrenal medulla, iTRAQ labels 114-117 5779 protein identified, 4414 with 2 or more peptides, FDR 0.01 Coverage # Unique Accession Description [%] Peptides Q9C0C9 (E3-independent) E2 ubiquitin-conjugating enzyme OS=Homo sapiens GN=UBE2O PE=1 SV=3 7 7 O14874 [3-methyl-2-oxobutanoate dehydrogenase [lipoamide]] kinase, mitochondrial OS=Homo sapiens GN=BCKDK PE=1 SV=2 7 2 Q8TDZ2 [F-actin]-monooxygenase MICAL1 OS=Homo sapiens GN=MICAL1 PE=1 SV=2 11 5 Q7RTP6 [F-actin]-monooxygenase MICAL3 OS=Homo sapiens GN=MICAL3 PE=1 SV=2 1 1 Q15118 [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 1, mitochondrial OS=Homo sapiens GN=PDK1 PE=1 SV=1 17 4 Q15119 [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 2, mitochondrial OS=Homo sapiens GN=PDK2 PE=1 SV=2 16 3 Q15120 [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 3, mitochondrial OS=Homo sapiens GN=PDK3 PE=1 SV=1 13 3 Q9P0J1 [Pyruvate dehydrogenase [acetyl-transferring]]-phosphatase 1, mitochondrial OS=Homo sapiens GN=PDP1 PE=1 SV=3 29 10 Q04446 1,4-alpha-glucan-branching enzyme OS=Homo sapiens GN=GBE1 PE=1 SV=3 38 19 P61604 10 kDa heat shock protein, mitochondrial OS=Homo sapiens GN=HSPE1 PE=1 SV=2 77 10 Q15029 116 kDa U5 small nuclear ribonucleoprotein component OS=Homo sapiens GN=EFTUD2 PE=1 SV=1 32 23 Q9NRX4 14 kDa phosphohistidine phosphatase OS=Homo sapiens GN=PHPT1 PE=1 SV=1 38 3 P31946 14-3-3 protein beta/alpha OS=Homo sapiens GN=YWHAB PE=1 SV=3 65 7 P62258 14-3-3 protein epsilon OS=Homo -

GPRASP/ARMCX Protein Family: Potential Involvement in Health And
GPRASP/ARMCX protein family: potential involvement in health and diseases revealed by their novel interacting partners Juliette Kaeffer, Gabrielle Zeder-Lutz, Frederic Simonin, Sandra Lecat To cite this version: Juliette Kaeffer, Gabrielle Zeder-Lutz, Frederic Simonin, Sandra Lecat. GPRASP/ARMCX pro- tein family: potential involvement in health and diseases revealed by their novel interacting part- ners. Current Topics in Medicinal Chemistry, Bentham Science Publishers, 2021, 21 (3), pp.227-254. 10.2174/1568026620666201202102448. hal-03095663 HAL Id: hal-03095663 https://hal.archives-ouvertes.fr/hal-03095663 Submitted on 4 Jan 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. GPRASP/ARMCX protein family: potential involvement in health and diseases revealed by their novel interacting partners Juliette Kaeffer, Gabrielle Zeder-Lutz, Frédéric Simonin* and Sandra Lecat* Affiliation : Biotechnologie et Signalisation Cellulaire, UMR7242 CNRS, Université de Strasbourg, Illkirch-Graffenstaden, France. * Corresponding authors: Sandra Lecat ([email protected]) and Frédéric Simonin ([email protected]) : Biotechnologie et Signalisation Cellulaire, UMR7242 CNRS / Université de Strasbourg, 300 boulevard Sébastien Brant, CS 10413, 67412 Illkirch-Graffenstaden, Cedex, France. 1 Abstract GPRASP (GPCR-associated sorting protein)/ARMCX (ARMadillo repeat-Containing proteins on the X chromosome) family is composed of 10 proteins, which genes are located on a small locus of the X chromosome except one.