Mechanism of Acid-Catalyzed Epoxidation of Alkenes with Peroxy Acids
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lab Safety Manual and the Laboratory-Specific Information in the CHP Easily
Laboratory Safety Manual With the Lab-Specific Chemical Hygiene Plan Template The University of Tennessee Knoxville Campus September 2015 PURPOSE OF THE LABORATORY SAFETY MANUAL 1) Provide laboratories with useful recommendations that can help achieve compliance with the OSHA Lab Standard and workplace safety rules and regulations. 2) Equip researchers to develop a Lab-Specific Chemical Hygiene Plan Throughout this document, areas where regulatory or University requirements exist will be clearly identified using words such as “must”, “required”, “shall”, and “it is the responsibility”, etc. All other information provided within this document serves as recommendations that Environmental Health and Safety (EHS) encourages laboratories to follow as best management practices. To take advantage of the Internet, this document is formatted to be a “front door” to other resources, including useful web links. Where appropriate, web links are provided that can be clicked to view the webpage. This Laboratory Safety Manual should be considered a living document and will be reviewed at least annually and updated with your participation, comments, and suggestions. Policy Statement It is the policy of the University of Tennessee at Knoxville (UTK) to provide a safe working and learning environment. The Environmental, Health, and Safety Department has developed this manual as a guidance document to familiarize UTK faculty, staff, students, volunteers, and visitors with the campus-wide policies and procedures for the safe use of hazardous chemicals and other hazardous materials at the University. When these policies and procedures are followed, the risk of occupational exposures to chemicals and physical hazards as well as the risk of accidental environmental release of hazardous materials is minimized. -

Changxia Yuan Baran Group Meeting 4/5/2014 Commercial Available
Baran Group Meeting Changxia Yuan 4/5/2014 Commercial available peroxides* Inorganic peroxides Na2O2 CaO2 Li2O2 BaO2 Ni2O3 NiO2 xH2O H2O2(30%) ZnO2 NaBO3 4H2O MgO2 TbO2 SrO2 Na2CO3 1.5H2O sodium calcium lithium barium nickel nickel(II) hydrogen zinc sodium magnesium terbium Stronium sodium peroxide peroxide peroxide peroxide peroxide peroxide peroxide peroxide perborate peroxide peroxide peroxide percarbonate $ 109/100g $ 27/100g $ 32 /50g $ 146/500g $ 106/5g hydrate $ 350/4L $ 75/1kg tetrahydrate complex $ 30/1g $ 38/100g $ 91/ 2.5kg $ 40/ 1g $94/1kg $ 40/250g + (NH4)2S2O8 Na2S2O8 K2S2O8 2K2SO5 KHSO4 K2SO4 5[Bu4N ] SO5] HSO4] SO4] ammonium sodium potassium Oxone® OXONE® persulfate persulfate persulfate monopersulfate tetrabutylammonium salt $ 39/ 100g $ 87/ 2.5kg $ 70/ 500g compound $ 156/ 25g $ 60/ 1kg Organic peroxides-1 O CO3H O HOO tBu O O OO O CH2(CH2)9CH3 O H3C(H2C)9H2C O O tBu tBuOOH urea H2O2 BzOOBz OOH O Cl O tert-Butyl Urea Benzoyl mCPBA Cyclobutane 2-Butanone tert-Butyl Lauoyl hydroperoxide hydrogen peroxide $ 81/100g maloyl peroxide peroxide peroxide solution (5-6 M) peroxide $ 92/500g peroxide $ 129/500mL $ 134/1L $ 81/100g $ 47/25mL $ 88/ 250g $ 100/1g Cl O O Cl O Me Me O Me Me Me Me O O O O Ph O O O O OO O Me O Me O Ph O O tBu Cl Ph Me Me Me O Me 2,4-Dichlorobenzoyl Cl tert-Butyl tert-Butyl peracetate solution, Dicumyl tert-Butylperoxy 1,1-Bis(tert-amylperoxy)cyclohexane peroxide, 50% in DBP peroxybenzoate 50% in mineral spirits peroxide 2-ethylhexyl solution, 80% in mineral spirits $ 59/100g $ 86/500mL $ 77/500mL $ 123/500g -

Towards the Synthesis of the Emestrin Family of Natural Products
Towards the Synthesis of the Emestrin Family of Natural Products Brendan James Fisher ORCID: 0000-0003-0090-8951 Submitted in total fulfilment of the requirements of the degree of Doctor of Philosophy September 2018 School of Chemistry Bio21 Institute The University of Melbourne Abstract A Cope rearrangement of a vinyl pyrrole epoxide (397) was utilised to form the dihydrooxepino[4,3-b]pyrrole core (398) of the emestrin family of natural products which involved the first examples of the dearomatisation of pyrrole in this type of rearrangement. It was found that an electron withdrawing ester substituent on the C2 position of the epoxide was essential for the [3,3]-rearrangement to occur. The vinyl pyrrole epoxides were synthesised in an efficient manner by a vinylogous Darzens reaction. Density functional calculations showed lower transition state energies for Cope rearrangements of epoxides with C2 esters when compared to the unsubstituted substrates which agreed with the observed experimental results. Silyl substituted vinyl bromide esters also participated in the Darzens reactions to give the desired vinyl pyrrole epoxides in good to excellent yields. Only the triethoxysilyl vinyl epoxide 313c underwent Cope rearrangement to provide the fully substituted emestrin core dihydrooxepine. The anion derived from an aryl bromosulfone did not give the Darzens product but underwent a previously unobserved stereoselective trimerization to afford the cyclohexene 343 as a single diastereoisomer. A mechanistic rationale involving SN2’ additions, [3,3]-Cope rearrangements and a stereoselective intramolecular conjugate addition was proposed and this was supported by density functional theory (DFT) calculations. A four-step total synthesis of biaryl ether natural product violaceic acid (11) is described. -

The Role of Acid Catalysis in the Baeyer−Villiger Reaction. a Theoretical Study Robert D
Article pubs.acs.org/joc The Role of Acid Catalysis in the Baeyer−Villiger Reaction. A Theoretical Study Robert D. Bach* Department of Chemistry and Biochemistry, University of Delaware, Newark, Delaware 19716, United States *S Supporting Information ABSTRACT: Quantum mechanical calculations at the B3LYP/6- 311+G(d,p) level have examined the overall mechanism of the Baeyer−Villiger (BV) reaction with peroxyacetic acid. A series of reactions that include both the addition step and the subsequent alkyl group migration step included ketones, acetone, t-butyl methyl ketone, acetophenone, cyclohexyl methyl ketone, and cyclohexyl phenyl ketone. The combined data suggested that the first step for addition of the peroxyacetic acid oxidation catalyst to the ketone carbonyl to produce the Criegee or tetrahedral intermediate is rate- limiting and has activation barriers that range from 38 to 41 kcal/ mol without the aid of a catalyst. The rate of addition is markedly reduced by the catalytic action of a COOH functionality acting as a donor−acceptor group affecting both its proton transfer to the ketone CO oxygen in concert with transfer of the OOH proton to the carboxylic acid carbonyl. The second or alkyl group migration step has a much reduced activation barrier, and its rate is not markedly influenced by acid catalysis. The rate of both steps in the BV reaction is greatly influenced by the catalytic action of very strong acids. ■ INTRODUCTION mechanism for addition to the carbonyl group and how the hemiperacetal hydrogen migrates to the carboxylate to produce The Baeyer−Villiger (BV) reaction remains an important the final products, an ester functionality and a carboxylic acid. -

Reaction-Of-Alkenes.Pdf
8 Reactions of Alkenes Goals for Chapter 8 1 Explain why electrophilic additions are among the most common reactions of alkenes. femoral component 2 Predict the products of the reactions of alkenes, including the orientation of the reaction (regio- polyethylene bearing chemistry) and the stereochemistry. tibial plate 3 Propose mechanisms to explain the observed products of alkene reactions. 4 Use retrosynthetic analysis to solve multistep synthesis problems with alkenes as reagents, intermediates, or products. ◀ Polyethylene provides a self-lubricating surface for movement of the metal parts of an artificial knee replacement. The highly cross-linked polyethylene used in these implants is fabricated to be exceptionally tough: It wears only about 0.1 mm per year. Polyethylene is compatible with the human body, and in most cases, it does not cause a foreign body reaction even after years of constant movement in the joint. The alkene double bond is a gateway functional group. Alkene reactions lead to many other functional groups that lay the foundation for the rest of your study of organic chemistry. You can convert alkenes to alkyl halides, epoxides, alcohols, aldehydes, ketones, carboxylic acids, and other functional groups. The reactions of alkenes arise from the reactivity of their carbon–carbon double bonds. Organic chemists enjoy the challenge of taking a simple carbon–carbon double bond and manipulating it in all possible ways to produce other compounds, often mimicking biological reactions that occur in cells. This chapter covers the most common alkene reactions, including their mechanisms, reactivity, orientation, and stereochemistry. Most reactions of alkenes involve addition of atoms or groups across the double bond, with one atom or group adding to each end. -

Reactions of Alkenes
Reactions of Alkenes Alkenes generally react in an addition mechanism (addition – two new species add to a molecule and none leave) R X Y X Y H R R R H Have already observed using a H+ electrophile (HBr or H+/H2O) that a carbocation intermediate is generated which directs the regiochemistry Whenever a free carbocation intermediate is generated there will not be a stereopreference due to the nucleophile being able to react on either lobe of the carbocation (already observed this with SN1 and E1 reactions) Br Br H+ H H3C Br CH2CH3 Obtain racemic mixture of this regioisomer Reactions of Alkenes There are three questions to ask for any addition reaction R X Y X Y H R R R H 1) What is being added? (what is the electrophile?) 2) What is the regiochemistry? (do the reagents add with the X group to the left or right?) 3) What is the stereochemistry? (do both the X and Y groups add to the same side of the double bond or opposite sides?) All of these questions can be answered if the intermediate structure is known Reactions of Alkenes Dihalogen compounds can also react as electrophiles in reactions with alkenes Possible partial bond structures + !+ Br Br ! Br Br Br !+ or Br !+ More stable partial positive charge Experimentally it is known, however, that rearrangements do nor occur with Br2 addition -therefore free carbocations must not be present The large size and polarizability of the halogen can stabilize the unstable carbocation With an unsymmetrical alkene, however, both bonds to the bromine need not be equivalent Called a “Bromonium” ion -

The Epoxidation of Olefins by Peroxy Compounds
THE EPOXIDATION OF OLEFINS BY PEROXY COMPOUNDS BY ALFREDO LUIS MARTINS LAMEIRAO MATEUS A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY UNIVERSITY OF FLORIDA 1998 This dissertation is dedicated to my wife, Deise Prina Dutra, without whom I would never have started or completed it. It is also dedicated to my parents, Mario Joaquim Mateus and Emilia Maria Martins Mateus who always supported me and to the memory of Dr. Russell S. Drago, Doc, who was a source of continued enthusiasm for this work. ACKNOWLEDGMENTS Dr. Russell Drago was much more than an adviser for me. I would like to express my deep admiration and respect for him in every possible way. He was a brilliant scientist and an esteemed friend. He welcomed me and the other members of the group into his family and into the big family of Drago Group members. This was of fundamental importance to me in adjusting to living in a different country. I hope someday I will be able to live up to his expectations and to do "his" reactions. I wish to thank Mrs. Ruth Drago for the hospitality and all the good times at the Dragos house. I am very grateful to everybody in the Drago Group during the years I spent here. The people in the oxidation sub-group contributed a lot, with ideas and discussion about all of my "4000 projects a day." I would like to thank Dr. Ken Lo, Cheng Xu, Ben Gordon, Karen Frank and the many undergraduate students and visiting scientists that came to work in our sub-group. -

Catalytic Developments in the Epoxidation of Vegetable Oils and the Analysis Methods of Cite This: RSC Adv.,2019,9,38119 Epoxidized Products
RSC Advances View Article Online REVIEW View Journal | View Issue Catalytic developments in the epoxidation of vegetable oils and the analysis methods of Cite this: RSC Adv.,2019,9,38119 epoxidized products Phyu Thin Wai, Pingping Jiang, * Yirui Shen, Pingbo Zhang, Qian Gu and Yan Leng Functionalization of vegetable oils (VOs) including edible, non-edible, and waste cooking oil (WCOs) to epoxides (EVOs) is receiving great attention by many researchers from academia and industry because they are renewable, versatile, sustainable, non-toxic, and eco-friendly, and they can partially or totally replace harmful phthalate plasticizers. The epoxidation of VOs on an industrial scale has already been developed by the homogeneous catalytic system using peracids. Due to the drawbacks of this method, other systems including acidic ion exchange resins, polyoxometalates, and enzymes are becoming alternative catalysts for the epoxidation reaction. We have reviewed all these catalytic systems including Received 31st July 2019 their benefits and drawbacks, reaction mechanisms, intensification of each system in different ways as Accepted 9th October 2019 Creative Commons Attribution-NonCommercial 3.0 Unported Licence. well as the physicochemical properties of VOs and EVOs and new findings in recent years. Finally, the DOI: 10.1039/c9ra05943a current methods including titrimetric methods as well as ATR-FTIR and 1H NMR for determination of rsc.li/rsc-advances conversion, epoxidation, and selectivity of epoxidized vegetable oils (EVOs) are also briefly described. 1. Introduction controlled plasticizer (along with DBP and BBP). In March 2015, the European Commission added DEHP/DOP as well as di Although phthalate plasticizers have been used for many normal butyl phthalate (DBP), diisobutyl phthalate (DIBP), and decades in poly(vinyl chloride) (PVC) insulation and jacketing of butylbenzyl phthalate (BBP) to Annex II of RoHS. -

1 Chapter 17 Ethers, Epoxides, Sulfides Ethers Are Much Less
Chapter 17 Ethers, Epoxides, Sulfides Ethers are much less reactive than alcohols but epoxides – three-membered ring ethers – are very reactive as we saw in the last chapter. Nomenclature In substitutive IUPAC nomenclature ethers are named as alkoxy derivative derivatives of alkanes. Functional class names: list the two alkyl groups in ROR’ in alphabetical order as separate words followed by the word “ether”. If the two alkyl groups are the same, use “di-“. Ethers can be symmetrical or unsymmetrical. In unsymmetrical ethers, the two alkyl groups are different. We can have cyclic ethers: Many compounds have more than one ether linkage. For example, some diethers are useful solvents and there are useful solvents that contain multiple ethers. These have higher boiling points and more ether linkages can be added to increase the boiling point even further. Sulfur analogs of alkoxy groups are called alkylthio groups. Name as sulfides or alkyl thio alkanes. 1 Structure and Bonding Ethers have large bond angles than water and alcohols due to van der Waals strain. Typical C-O bond lengths are similar to C-O bonds in alcohols (~1.42 A°). These are shorter than typical C-C bonds (~1.52A°) The most stable conformation of diethyl ether is the all-staggered anti-conformation. Tetrahydropyran is most stable in the chair conformation. In a three-membered ring the bond angles are much small than normal tetrahedral angles and the C-C bond and C-O are slightly longer than normal due to the severe angle strain. Physical Properties Look at diethyl ether as compared to pentane and 1-butanol. -

Lecture Notes Chem 51B S. King Chapter 12 Oxidation & Reduction I
Lecture Notes Chem 51B S. King Chapter 12 Oxidation & Reduction I. Introduction In this chapter we will discuss the oxidation and reduction of akenes, alkynes, alcohols, ethers, and epoxides. Oxidation & reduction reactions are very valuable in organic synthesis. Recognizing whether a compound is oxidized or reduced is an important first step in being able to successfully choose the correct reagents in a chemical transformation. A. Recognizing Oxidation and Reduction of Organic Compounds Oxidation-reduction reactions involve the gain and loss of electrons, and a change in the oxidation state of the molecules involved: Oxidation: loss of electrons Reduction: gain of electrons Cu + 2Ag+ Cu2+ + Ag shiny red metal colorless blue silver crystals If one molecule is oxidized, another is reduced. Electrons are TRANSFERRED COMPLETELY from one molecule to another. For organic compounds, oxidation-reduction reactions result in a CHANGE IN ELECTRON DENSITY around the carbon atom rather than a complete transfer of electrons. Oxidation: loss of "electron density" Carbon "loses" electrons by forming bonds with elements that are more electronegative than it is. loss of: gain of: 87 Reduction: gain of "electron density" Carbon "gains" electrons by giving up bonds to more electronegative elements and forming bonds with hydrogen atoms instead. loss of: gain of: The following shows stepwise oxidation of methane (the most reduced form of carbon) all the way up to carbon dioxide (the most oxidized form of carbon): H H H H H C H HO C H C O C O O C O H H H HO Can you recognize oxidation and reduction in the following examples? O O C CH2CH2OH CH3 OH H C H H3C H 3 H2O C C H C C H H O+ H H 3 HO H 88 II. -

Lb Transition Structure for the Epoxidation of Alkenes with Peroxy
2338 J. Am. Chem. SOC.1991, 113, 2338-2339 be of the order of lo3 M-l at 298 K when B is 2,6-dimethyl- than that found for hydrogen peroxide.' pyridine.14 Since the hydrogen-bonding equilibrium is expected We first examined the relative energies of the two zwitterionic to be exceedingly rapid, these data support the interpretation species arising from 1,2- and 1,4-hydrogen shifts in peroxyformic presented in the previous paragraph for the rapid proton-transfer acid (1). Dioxygen ylide la and hydroxycarbonyl oxide lb are reactions. The second-order rate constant (Table 11) for the reaction of OjH1 PA-NHz'+ with TBP exceeds that for the transfer of a benzylic proton from PA-CH3'+ to TBP by more than lo4 even though 01 - c Oi the latter reaction is thermodynamically more favorable by a factor H2/ \+/ + H~/'\O< 02 of 1 OI4. A similar comparison for the reactions of PA-H'+ and lb PA-CHl'+ with TBP reveals that the former reacts 5 times as fast 1 la A, in spitebf the fact that the equilibrium constant is 14 orders of calculated to be 55.5 and 28.8 kcal/mol higher in energy than magnitude less favorable. This apparent inverse dependence of 1, but the barrier for reversion of lb to 1 is only 1.7 kcal/mol kinetic on thermodynamic acidities further illustrates the com- (HF/6-31G*).* As in HOOH H200,the barrier disappears plexities of cation radical deprotonation reactions. Likewise, there - when MP4 correlation corrections are incl~ded.~With geometry is no direct relationship between cation radical reduction potential optimization at the MP2/6-31G* level, water oxide does exist as and kinetic acidities in this series of reactions. -
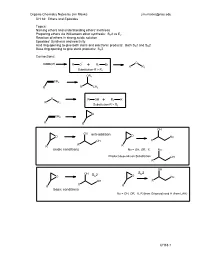
Acidic Conditions Anti-Addition Basic Conditions S 2
Organic Chemistry Notes by Jim Maxka [email protected] CH 18: Ethers and Epoxides Topics: Naming ethers and understanding ethers’ inertness Preparing ethers via Williamson ether synthesis: SN2 vs E2 Reaction of ethers in strong acidic solution Epoxides’ Synthesis and reactivity Acid ring opening to give both steric and electronic products: Both SN1 and SN2 Base ring opening to give steric products: SN2 Connections: ROH - O RO+ R1 Br R R1 Substitution-R > R1 OR1 CH2 R RCH3 O ROH+ R1 X R R1 Substitution-R > R1 O CH2 R R OH OH anti-addition O O Nu OH R R R R acidic conditions Nu = OH, OR, X Nu Product depends on Substitution OH R OH S 2 OH S 2 N O N O Nu OH R R R R basic conditions Nu = OH, OR, X, R (from Grignard) and H (from LAH) CH18-1 Organic Chemistry Notes by Jim Maxka [email protected] Ethers and Epoxides Naming ethers: Common: Take the substituent names of each side of the O and add ether Name: CH3CH2OCH2CH3 (CH3)2CHOCH2Ph (CH3)3COCH3 IUPAC: Take the shortest chain and make it a substituent with O (alkoxy)alkane. Name the above. Epoxides are cyclic ethers base on a 3-membered ring. Since epoxides are made from alkenes, we usually call them by the alkene name followed by “oxide”. The IUPAC name is epoxy denoting the positions. (e.g. 1,2-epoxy-) O O O O Physical properties: Why does ethanol boil at 780 while diethyl ether boils near room temperature? Why is ethanol completely soluble in water and boils as a 95:5 mixture with water, a while diethyl ether is used as an extraction solvent with water? b When mixed with water, ether separates by more than 97% (there is a tiny solubility – Do you know why? What technique must we use to deal with the solubility of ether in water after the extraction?) When ether is mixed with water does it go to the top or bottom? How can you tell? Assume similar volume for the unit of an organic (CH2) vs OH2.