Compounding Sterile Preparations, 4Th Edition Chapter 4
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Management of Poisoning
MOH CLINICAL PRACTICE GUIDELINES December/2011 Management of Poisoning Health Ministry of Sciences Chapter of Emergency College of College of Family Manpower Authority Physicians Physicians, Physicians Academy of Medicine, Singapore Singapore Singapore Singapore Medical Pharmaceutical Society Society for Emergency Toxicology Singapore Paediatric Association of Singapore Medicine in Singapore Society (Singapore) Society Executive summary of recommendations Details of recommendations can be found in the main text at the pages indicated. Principles of management of acute poisoning – resuscitating the poisoned patient GPP In a critically poisoned patient, measures beyond standard resuscitative protocol like those listed above need to be implemented and a specialist experienced in poisoning management should be consulted (pg 55). GPP D Prolonged resuscitation should be attempted in drug-induced cardiac arrest (pg 55). Grade D, Level 3 1 C Titrated doses of naloxone, together with bag-valve-mask ventilation, should be administered for suspected opioid-induced coma, prior to intubation for respiratory insuffi ciency (pg 56). Grade C, Level 2+ D In bradycardia due to calcium channel or beta-blocker toxicity that is refractory to conventional vasopressor therapy, intravenous calcium, glucagon or insulin should be used (pg 57). Grade D, Level 3 B Patients with actual or potential life threatening cardiac arrhythmia, hyperkalaemia or rapidly progressive toxicity from digoxin poisoning should be treated with digoxin-specifi c antibodies (pg 57). Grade B, Level 2++ B Titrated doses of benzodiazepine should be given in hyperadrenergic- induced tachycardia states resulting from poisoning (pg 57). Grade B, Level 1+ D Non-selective beta-blockers, like propranolol, should be avoided in stimulant toxicity as unopposed alpha agonism may worsen accompanying hypertension (pg 57). -

Synthesis, Physicochemical Characterization and Study of the Antimicrobial Activity of Chlorobutanol H
World Academy of Science, Engineering and Technology International Journal of Pharmacological and Pharmaceutical Sciences Vol:12, No:3, 2018 Synthesis, Physicochemical Characterization and Study of the Antimicrobial Activity of Chlorobutanol H. Nadia, G. Bahdja, S. Thili Malha, Y. Zahoua, D. Taoufik, B. Mourad, M. Marzouk, F. Z. Hadjadj Aoul, L. R. Mekacher I. INTRODUCTION Abstract—Introduction and objectives: Chlorobutanol is a raw XCIPIENTS play a vital role in the design, manufacture material, mainly used as an antiseptic and antimicrobial preservative and preservation of drugs. The addition of an in injectable and ophthalmic preparations. The main objective of our E study was the synthesis and evaluation of the antimicrobial activity of antimicrobial agents is of great interest in the case where the chlorobutanol hemihydrates. Material and methods: Chlorobutanol pharmaceutical preparations do not have adequate was synthesized according to the nucleophilic addition reaction of antimicrobial properties, (especially aqueous preparations), to chloroform to acetone, identified by an infrared absorption using prevent proliferation or limit microbial contamination [1], [2]. Spectrum One FTIR spectrometer, melting point, Scanning electron Chlorobutanol is a trihalogen alcohol with bacteriostatic and microscopy and colorimetric reactions. The dosage of Carvedilol fungistatic activity. The main objective of our work was to active substance was carried out by assaying the degradation products of chlorobutanol in a basic solution. The chlorobutanol obtained was obtain chlorobutanol hemihydrate by chemical synthesis, its subjected to bacteriological tests in order to study its antimicrobial purification and characterization as well as the study of its activity. The antibacterial activity was evaluated against strains such antimicrobial activity. as Escherichia coli (ATCC 25 922), Staphylococcus aureus (ATCC 25 923) and Pseudomonas aeroginosa (ATCC = American type II. -

GHS Calcium Lactate Gluconate MSDS.Pdf
Safety Data Sheet (Calcium Lactate Gluconate) DATE PREPARED: 7/9/2015 Section 1. Product and Company Identification Product Name Calcium Lactate Gluconate CAS Number 11116-97-5 Parchem - fine & specialty chemicals EMERGENCY RESPONSE NUMBER 415 Huguenot Street CHEMTEL New Rochelle, NY 10801 Toll Free US & Canada: 1 (800) 255-3924 (914) 654-6800 (914) 654-6899 All other Origins: 1 (813) 248-0585 parchem.com [email protected] Collect Calls Accepted Section 2. Hazards Identification Classification of the substance or mixture Classification according to Directive 67/548/EEC or 1999/45/EC as amended: This preparation does not meet the criteria for classification according to Directive 1999/45/EC as amended. Classification according to Regulation (EC) No 1272/2008 as amended: This mixture does not meet the criteria for classification according to Regulation (EC) 1272/2008 as amended. Hazard and precautionary statements Hazard statements: The substance does not meet the criteria for classification. Precautionary statements: Not available. Appearance & Odor: White powder with no odor. Fire & Explosion Hazards Potential for dust explosion may exist. This product is not defined as flammable or combustible. However, the product may decompose under fire conditions to produce toxic oxides of carbon. Depending upon conditions, dusts may be sensitive to static discharge. Avoid possibility of dry powder and friction causing static electricity in presence of flammables (See NFPA-77, Chpt. 6) Primary Route of Exposure: Skin and eye contact are the primary routes of exposure to this product. Inhalation Acute Exposure: Inhalation of dust may cause mild irritation. Skin Contact - Acute: Skin contact is not expected to cause irritation. -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

Wednesday May 26, 1999
5±26±99 Vol. 64 No. 101 Wednesday Pages 28333±28712 May 26, 1999 federal register 1 VerDate 06-MAY-99 21:29 May 25, 1999 Jkt 183247 PO 00000 Frm 00001 Fmt 4710 Sfmt 4710 E:\FR\FM\26MYWS.XXX pfrm03 PsN: 26MYWS II Federal Register / Vol. 64, No. 101 / Wednesday, May 26, 1999 The FEDERAL REGISTER is published daily, Monday through SUBSCRIPTIONS AND COPIES Friday, except official holidays, by the Office of the Federal Register, National Archives and Records Administration, PUBLIC Washington, DC 20408, under the Federal Register Act (44 U.S.C. Subscriptions: Ch. 15) and the regulations of the Administrative Committee of Paper or fiche 202±512±1800 the Federal Register (1 CFR Ch. I). The Superintendent of Assistance with public subscriptions 512±1806 Documents, U.S. Government Printing Office, Washington, DC 20402 is the exclusive distributor of the official edition. General online information 202±512±1530; 1±888±293±6498 Single copies/back copies: The Federal Register provides a uniform system for making available to the public regulations and legal notices issued by Paper or fiche 512±1800 Federal agencies. These include Presidential proclamations and Assistance with public single copies 512±1803 Executive Orders, Federal agency documents having general FEDERAL AGENCIES applicability and legal effect, documents required to be published Subscriptions: by act of Congress, and other Federal agency documents of public Paper or fiche 523±5243 interest. Assistance with Federal agency subscriptions 523±5243 Documents are on file for public inspection in the Office of the Federal Register the day before they are published, unless the issuing agency requests earlier filing. -

CAS No.) Screening Justification Is Public Comment Now Open? CAS No
EGLE Air Quality Division (AQD) Air Toxics Screening Level Justifications (Numerically by CAS No.) Screening Justification Is Public Comment Now Open? CAS No. Chemical Name Level & Responses Info E-Mail AQD Deadline None ad acid View View Not Open for Public Comment None amyl acetate (mixture) View View Not Open for Public Comment None atlox 848 View View Not Open for Public Comment None biosam tp-1.5 View View Not Open for Public Comment None calcium chloride View View Not Open for Public Comment None epoxy resin solution View View Not Open for Public Comment None heptamethyl-1-vinyl-1,7-dichlorotetrasilazane View View Not Open for Public Comment None n-butylglucamine View View Not Open for Public Comment None n-chloro-2,6-difluorobenzamide View View Not Open for Public Comment None trichloroethylene View View Not Open for Public Comment None triethylammonium suleptanate View View Not Open for Public Comment 50-00-0 formaldehyde View View Not Open for Public Comment 50-03-3 hydrocortisone acetate View View Not Open for Public Comment 50-21-5 lactic acid View View Not Open for Public Comment 50-28-2 estradiol View View Not Open for Public Comment 50-29-3 ddt View View Not Open for Public Comment 50-32-8 benzo(a)pyrene View View Not Open for Public Comment 51-28-5 2,4-dinitrophenol View View Not Open for Public Comment 53-36-1 methyl predisolone acetate View View Not Open for Public Comment 53-70-3 dibenz(a,h)anthracene View View Not Open for Public Comment 56-23-5 carbon tetrachloride View View Not Open for Public Comment 56-49-5 3-methylcholanthrene View View Not Open for Public Comment 56-55-3 benz(a)anthracene View View Not Open for Public Comment 56-81-5 glycerol View View Not Open for Public Comment 57-11-4 stearic acid View View Not Open for Public Comment Revised Monday, September 27, 2021 Page 1 of 51 EGLE Air Quality Division (AQD) Air Toxics Screening Level Justifications (Numerically by CAS No.) Screening Justification Is Public Comment Now Open? CAS No. -

Guide to Extractables in Effluents from Ultipor® N66 and Posidyne® Filter
Validation Guide USTR 2521 Guide to Extractables in Effluents from Pall Ultipor ® N66 and Posidyne ® Filter Cartridges Contents 1. Introduction ........................................................................................................................3 2. Design and Manufacture of Pall N66 Pharmaceutical-rated (P-rated) Filters ................3 3. Principles of Extraction and the Non-Volatile Residue Test ............................................3 3.1 Factors Limiting Extraction ............................................................................................3 3.2 Defining Solvent Strength ..............................................................................................4 3.3 Materials of Construction of Ultipor N66 and Posidyne Filter Cartridges ........................5 3.4 Extractable Substances in Polymers Used in Pall Ultipor N66 and Posidyne Filter Cartridges ....................................................................................................5 4. Total Non-volatile Residues Extracted from Ultipor N66 and Posidyne Filters ..............6 4.1 Introduction ....................................................................................................................6 4.2 Summary of Method ......................................................................................................6 4.3 Results ..........................................................................................................................6 4.4 Conclusions ..................................................................................................................6 -

Calcium Gluconate
CALCIUM GLUCONATE CLASSIFICATION Minerals and electrolytes TRADE NAME(S) Calcium Gluconate 10% (for IV use) Calcium Gluconate gel 2.5% (for topical use) DESIRED EFFECTS Lower potassium levels; pain relief and neutralizing fluoride ion MECHANISM OF ACTION Calcium is the primary component of skeletal tissue. Bone serves as a calcium depot and as a reservoir for electrolytes and buffers. INDICATIONS Suspected hyperkalemia in adult PEA/Asystole Antidote for calcium channel blocker overdose and magnesium sulfate toxicity Gel is used for hydrofluoric acid burns Suspected hyperkalemia with adult crush injury or peaked T-waves on EKG CONTRAINDICATIONS Should not be given to patients with digitalis toxicity Should be used with caution in patients with dehydration ADVERSE REACTIONS When given too rapidly or to someone on digitalis, can cause sudden death from ventricular fibrillation May cause mild to severe IV site irritation SPECIAL CONSIDERATIONS Must either use a different IV line or flush line with copious normal saline if being given with sodium bicarbonate. When used on hydroflouric acid burns, relief of pain is the only indication of treatment efficacy. Therefore, use of analgesic agents is not recommended. DOSING REGIMEN Suspected hyperkalemia in adult PEA/asystole or adult crush injury, or evidence of EKG changes (Ex. peaked T-waves) o Calcium gluconate 10% 15-30 ml IV/IO over 2-5 minutes KNOWN calcium channel blocker overdose - administer 3 grams IV/IO may repeat dose in 10 minutes if no effect. Hydrofluoric acid burns apply calcium gluconate gel 2.5% every 15 minutes to burned area and massage continuously until pain disappears. -

United States Patent Office Patented Aug
2,760,990 United States Patent Office Patented Aug. 28, 1956 acid. If an acid group other than acetate is desired, mercury acetate and higher boiling carboxylic acid may 2,760,990 be intimately mixed together and the mixture is taken VNY TRANSETHERFICATION up with the starting vinyl ether. At this point the 5 mixture may be heated with stripping off of acetic acid. Warren H. Watanabe, Philadelphia, Pa., and Lawrence The reacting alcohol is then added and the mixture heated. E. Conlon, Moorestown, N.J., assignors to Rohm & Alternatively mercury acetate, carboxylic acid, vinyl Haas Company, Philadelphia, Pa., a corporation of ether, and alcohol are mixed and heated. Acetic acid Delaware may now be distilled off. The preparation of catalyst in No Drawing. Application August 15, 1952, O these ways, gives a highly active material, effective even Serial No. 304,648 at elevated temperatures. Another method of forming transetherifying catalysts 9 Claims. (C.260-614) is to mix together a common mercury salt of a strong acid, such as mercuric sulfate, and an alkali metal salt This invention concerns a process for preparing vinyl 5 of a desired carboxylic acid. Thus mercuric sulfate may ethers by transetherifying other vinyl ethers with alcohols be mixed with sodium oleate, or sodium benzoate, or in the presenec of soluble mercury salts of weak acids. It sodium ethylbutyrate. Such mixtures simulate the mer also deals with novel vinyl ethers, cury salts of weak acids, although they still give consid It has been proposed that volatile vinyl alkyl ethers erable acetal formation. in which the alkyl group is primary be reacted with pri 20 When alcohols are used which are acid sensitive, or mary or secondary monohydric alcohols: consisting of which are unstable to acid, or which decompose to give. -

Papaverine Hydrochloride Injection, Solution American Regent, Inc
PAPAVERINE HYDROCHLORIDE- papaverine hydrochloride injection, solution American Regent, Inc. Disclaimer: This drug has not been found by FDA to be safe and effective, and this labeling has not been approved by FDA. For further information about unapproved drugs, click here. ---------- PAPAVERINE HYDROCHLORIDE INJECTION, USP Rx Only This product is to be used by or under the direction of a physician. Each vial contains a sufficient amount to permit withdrawal and administration of the volume specified on the label. DESCRIPTION Papaverine Hydrochloride, USP, is the hydrochloride of an alkaloid obtained from opium or prepared synthetically. It belongs to the benzylisoquinoline group of alkaloids. It does not contain a phenanthrene group as do morphine and codeine. Papaverine Hydrochloride, USP, is 6,7-dimethoxy-1- veratrylisoquinoline hydrochloride and contains, on the dried basis, not less than 98.5% of C20H21NO4•HCI. The molecular weight is 375.85. The structural formula is as shown. Papaverine Hydrochloride occurs as white crystals or white crystalline powder. One gram dissolves in about 30 mL of water and in 120 mL of alcohol. It is soluble in chloroform and practically insoluble in ether. Papaverine Hydrochloride Injection, USP, is a clear, colorless to pale-yellow solution. Papaverine Hydrochloride, for parenteral administration, is a smooth-muscle relaxant that is available in vials containing 30 mg/mL. Each vial also contains edetate disodium 0.005%. The 10 mL vials also contain chlorobutanol 0.5% as a preservative. pH may be adjusted with sodium citrate and/or citric acid. CLINICAL PHARMACOLOGY The most characteristic effect of papaverine is relaxation of the tonus of all smooth muscle, especially when it has been spasmodically contracted. -
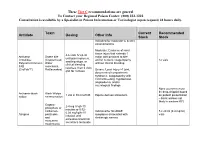
Antidote Stocking Tier C
These Tier C recommendations are general To Contact your Regional Poison Center: (800) 222-1222 Consultation is available by a Specialist in Poison Information or Toxicologist (upon request) 24 hours daily. Toxin Current Recommended Antidote Dosing Other Info Stock Stock Indicated for moderate to severe envenomations Moderate: Evidence of local tissue injury that extends 1 4-6 vials IV q2-4h Antivenin Snake bite major joint proximal to bite until pain improves, Crotalidae (Copperhead, and/or numeric coagulopathy 12 vials swelling stops, or Polyvalent Immune Water without clinical bleeding clinical bleeding FAB moccasins, resolves, then 2 vials (CroFab™) Rattlesnakes) Severe: Local injury >1 joint, q6h for 3 doses documented compartment syndrome, coagulopathy with clinical bleeding, hypotension, angioedema, and/or neurological findings None (currently must be drop-shipped based Antivenin-black Black Widow 1 vial in 50 ml of NS Equine derived antivenom on patient presentation widow envenomation – black widows not likely in western KY) Organo- 2-4 mg IV q5-10 phosphate or minutes or 0.02- carbamate Indicated for SLUDGE 5 x 20 ml (0.4 mg/ml) 0.08 mg/kg/hr IV Atropine pesticides symptoms associated with vials infusion until and cholinergic excess excessive bronchial muscarine secretions terminate mushrooms These Tier C recommendations are general To Contact your Regional Poison Center: (800) 222-1222 Consultation is available by a Specialist in Poison Information or Toxicologist (upon request) 24 hours daily. Antidote Current Recommended Toxin -

Calcium Gluconate Injection Contains 95 Mg/Ml Calcium Gluconate Monohydrate and 3.0 Mg/Ml Calcium Saccharate
Calcium Gluconate Injection Contains 95 mg/mL Calcium Gluconate Monohydrate and 3.0 mg/mL Calcium Saccharate Consumer Medicine Information What is in this leaflet Before you are given You should not be given Calcium Gluconate Injection if you have been Calcium Gluconate bed ridden for a long time causing This leaflet answers some common the loss of calcium from the bones. questions about Calcium Gluconate Injection Injection. It does not contain all the You should not be given Calcium available information. It does not take Gluconate Injection if you are being the place of talking to your doctor. When you must not be given treated with certain heart drugs such it as digoxin and digitalis. All medicines have risks and benefits. Your doctor has weighed the risks of You should not be given Calcium You should not be given Calcium you being given Calcium Gluconate Gluconate Injection if you have an Gluconate Injection if the solution is Injection against the benefits they allergy to: discoloured, cloudy, turbid, or a expect it will have for you. • any medicine containing calcium precipitate is present. The solution is gluconate normally a clear, colourless liquid. If you have any concerns about being given this medicine, ask your • any of the ingredients listed at the You should not be given Calcium doctor. end of this leaflet. Gluconate Injection if when diluted with another solution it causes the Some of the symptoms of an allergic Keep this leaflet. solution to precipitate, become You may need to read it again. reaction may include: cloudy, turbid, discolour, or particles • shortness of breath are visible.