Recruitment of LC3 to Damaged Golgi Apparatus
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metric System Units of Length
Math 0300 METRIC SYSTEM UNITS OF LENGTH Þ To convert units of length in the metric system of measurement The basic unit of length in the metric system is the meter. All units of length in the metric system are derived from the meter. The prefix “centi-“means one hundredth. 1 centimeter=1 one-hundredth of a meter kilo- = 1000 1 kilometer (km) = 1000 meters (m) hecto- = 100 1 hectometer (hm) = 100 m deca- = 10 1 decameter (dam) = 10 m 1 meter (m) = 1 m deci- = 0.1 1 decimeter (dm) = 0.1 m centi- = 0.01 1 centimeter (cm) = 0.01 m milli- = 0.001 1 millimeter (mm) = 0.001 m Conversion between units of length in the metric system involves moving the decimal point to the right or to the left. Listing the units in order from largest to smallest will indicate how many places to move the decimal point and in which direction. Example 1: To convert 4200 cm to meters, write the units in order from largest to smallest. km hm dam m dm cm mm Converting cm to m requires moving 4 2 . 0 0 2 positions to the left. Move the decimal point the same number of places and in the same direction (to the left). So 4200 cm = 42.00 m A metric measurement involving two units is customarily written in terms of one unit. Convert the smaller unit to the larger unit and then add. Example 2: To convert 8 km 32 m to kilometers First convert 32 m to kilometers. km hm dam m dm cm mm Converting m to km requires moving 0 . -

Gravity and Coulomb's
Gravity operates by the inverse square law (source Hyperphysics) A main objective in this lesson is that you understand the basic notion of “inverse square” relationships. There are a small number (perhaps less than 25) general paradigms of nature that if you make them part of your basic view of nature they will help you greatly in your understanding of how nature operates. Gravity is the weakest of the four fundamental forces, yet it is the dominant force in the universe for shaping the large-scale structure of galaxies, stars, etc. The gravitational force between two masses m1 and m2 is given by the relationship: This is often called the "universal law of gravitation" and G the universal gravitation constant. It is an example of an inverse square law force. The force is always attractive and acts along the line joining the centers of mass of the two masses. The forces on the two masses are equal in size but opposite in direction, obeying Newton's third law. You should notice that the universal gravitational constant is REALLY small so gravity is considered a very weak force. The gravity force has the same form as Coulomb's law for the forces between electric charges, i.e., it is an inverse square law force which depends upon the product of the two interacting sources. This led Einstein to start with the electromagnetic force and gravity as the first attempt to demonstrate the unification of the fundamental forces. It turns out that this was the wrong place to start, and that gravity will be the last of the forces to unify with the other three forces. -

The International Bureau of Weights and Measures 1875-1975
The International Bureau of Weights and Measures 1875-1975 U.S. DEPARTMENT OF COMMERCE National Bureau of Standards ""EAU of NBS SPECIAL PUBLICATION 420 Aerial view of the Pavilion de Breteuil and the immediate environs. To the east, the Seine and the Pont de Sevres; to the northwest, the Pare de Saint-Cloud: between the Pavilion de Breteuil (circled) and the bridge: the Manufacture Nationale de Porcelaine de Sevres. The new laboratories (1964) are situated north of the circle and are scarcely visible; they were built in a way to preserve the countryside. (Document Institute (leographique National, Paris). Medal commeiiKiraUn-i the centennial (if the Convention cif tlie Metre and the International Bureau of Weights and Measures. (Desifined by R. Corbin. Monnaie de Paris) The International Bureau of Weights and Measures 1875-1975 Edited by Chester H. Page National Bureau of Standards, U.S.A. and Pan I Vigoiireiix National Physieal Laboratory, U.K. Translation of tlie BIPM Centennial Volume Piibli>lieH on the ocrasioii <>( the lOOth Aniiiver^ai y ol tlie Treaty of tlie Metre May 20, 1975 U.S. DEPARTMENT OF COMMERCE NATIONAL BUREAU OF STANDARDS, Richard W. Roberts, Direcior Issued May 1 975 National Bureau of Standards Special Publication 420 Nat. Bur. Stand. (U.S.), Spec. Publ. 420. 256 pages (May 1975) CODEN: XNBSAV U.S. GOVERNMENT PRINTING OFFICE WASHINGTON: 1975 For sale by the Superintendent of Documents U.S. Government Printing Office, Washington, D.C. 20402 Paper cover Price $3.00 Stock Number 003-003-01408 Catalog Number C13.10:420 FOREWORD The metric system was made legal by Congress in 1866, the United States of America signed the Treaty of the Metre in 1875, and we have been active in international coordination of measurements since that time. -
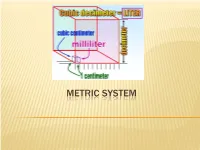
Metric System.Pdf
METRIC SYSTEM THE METRIC SYSTEM The metric system is much easier. All metric units are related by factors of 10. Nearly the entire world (95%), except the United States, now uses the metric system. Metric is used exclusively in science. Because the metric system uses units related by factors of ten and the types of units (distance, area, volume, mass) are simply-related, performing calculations with the metric system is much easier. METRIC CHART Prefix Symbol Factor Number Factor Word Kilo K 1,000 Thousand Hecto H 100 Hundred Deca Dk 10 Ten Base Unit Meter, gram, liter 1 One Deci D 0.1 Tenth Centi C 0.01 Hundredth Milli M 0.001 Thousandth The metric system has three units or bases. Meter – the basic unit used to measure length Gram – the basic unit used to measure weight Liter – the basic unit used to measure liquid capacity (think 2 Liter cokes!) The United States, Liberia and Burma (countries in black) have stuck with using the Imperial System of measurement. You can think of “the metric system” as a nickname for the International System of Units, or SI. HOW TO REMEMBER THE PREFIXES Kids Kilo Have Hecto Dropped Deca Over base unit (gram, liter, meter) Dead Deci Converting Centi Metrics Milli Large Units – Kilo (1000), Hecto (100), Deca (10) Small Units – Deci (0.1), Centi (0.01), Milli (0.001) Because you are dealing with multiples of ten, you do not have to calculate anything. All you have to do is move the decimal point, but you need to understand what you are doing when you move the decimal point. -

Rtusaeatz Nt,Rnasiivene
THE EVENING STAR. 38488 RQQe PUBLISRIED I AILY, Except Sunday, AT THE STAR BUILDINGS, any mznslo to sen e ear.te war to Avene, Corner 11th Street, 0011T the2ing f 1a0 tothi6 clany, it is Pensaylrania by partinenty as And why aboud as me '1he Evening Star Newspaper Oompany. King at sia1 reCeve the same alten as did 4:EQRUE W. ADAMS, Pres'r. one of his subjeCm alG the representative of a Tn EvrtNan Sra i. P.red to niscribers in the royal line, who, being aocredite to the paent r ty l. ,arr rP, on ti.eir own ateount, at 10 eents amInstmration, was, at the requet of Mr. 1 er w.ak or 44 eente per wonth. Colies at the Sicmes, our consul at Bangkok, sent from Siam eT:nt; r. - cnte each. by wail-sjotaKe prel aid- on one of our naval veeea, was received by toa eente a Llouth ; ene ye,r, $6; six months, I3. the iEterPd at the Poet Office t Washington, 0., President and Mrs. Hayes at the White -ase no. ca- mail ntatter 1 Housewith effusion, entertained there by them 'Ia V.1KT ST-r-p>b ished on F.atay-*2 a and mated asa member of thefamily for a con- errst trive irni;+i. Six months, 4; 10 copies siderable lenah of time? Every attention was for j1.: 20 col, . f.r $.. ARCH lavished on this myal guest; and should not the WAll nail ";bsCriltloine meit be paid in ad- CEN handsome young King receive as much? Yet It nee; > pa r ent l.ug than o paid for. -

Report to AID on a Philippines Survey on Standardization and Measurement Services
TECH NATL INST OF STAND & NIST PUBLICATIONS A111D? OSfilb^ IMBSIR 76-1083 Report to AID on a Philippines Survey on Standardization and Measurement Services Edited by: H. Steffen Peiser Robert S. Marvin Office of International Relations National Bureau of Standards Washington, D. C. 20234 Conducted May 4 17, 1975 Issued June 1 976 The Survey was conducted as a part of the program under the US/NBS/Agency for International Development PASA TA(CE) 5-71 \ epared for gency for International Development * 7L'/b83 epartment of State jCj^ Washington, D. C. 20523 NBSIR 76-1083 REPORT TO AID ON A PHILIPPINES SURVEY ON STANDARDIZATION AND MEASUREMENT SERVICES Edited by: H. Steffen Peiser Robert S. Marvin Office of International Relations National Bureau of Standards Washington, D. C. 20234 Conducted May 4 - 17, 1975 Issued June 1 976 The Survey was conducted as a part of the program under the US/NBS/Agency for International Development PASA TA(CE) 5-71 Prepared for Agency for International Development Department of State Washington, D. C. 20523 U.S. DEPARTMENT OF COMMERCE, Elliot L. Richardson, Secretary Dr. Betsy Ancfcer-Johrtsor At**st*nt Secretly for Science end Technology NATIONAL BUREAU OF STANDARDS. Ernest Ambler. Acting Director i TABLE OF CONTENTS Paee PARTICIPANTS 1 I INTRODUCTION 4 II RECOMMENDATIONS - SUMMARIZED 6 III THE JOINT PROGRAM OF THE SURVEY TEAM 12 IV REPORT OF GROUP A, TECHNICAL STANDARDS COMMITTEE MANAGEMENT, Group Leader; Dr. Kenneth S. Stephens, School of Industrial & Systems Engineering and Industrial Development Division, Engr. Exp. Station, Georgia Institute of Technology, Atlanta, Georgia 29 V REPORT OF GROUP B, METRICATION Group Leader; Mr. -

Enlightenment Bls.Qxd
Name _________________ All About the Enlightenment: The Age of Reason 1 Pre-Test Directions: Answer each of the following either True or False: 1. The leading figures of the Enlightenment era glorified reason (rational thought). ________ 2. Most of the main ideas put forth by the political philosophers of the Enlightenment era were rejected by the leaders of the American and French Revolutions. ________ 3. Electricity was studied during the Enlightenment era. ________ 4. Science during the Enlightenment era advanced more slowly than during the Renaissance. ________ 5. Deists held religious beliefs that were close to those of Catholics. ________ © 2004 Ancient Lights Educational Media Published and Distributed by United Learning All rights to print materials cleared for classroom duplication and distribution. Name _________________ All About the Enlightenment: The Age of Reason 2 Post-Test Fill in the blanks: 1. _______________ devised a system for classifying living things. 2. ______________ and______________ are credited with developing the "scientific method." 3. Laws of gravity and motion were formulated by _____________ in the 1660s. 4. The human ability to ___________ was glorified during the Enlightenment. 5. Anton Van Leeuwenhoek and Robert Hooke used _____________ in their studies. Essay Question: Name and discuss the contributions of the French and English philosophers of the Enlightenment to the development of American democracy. © 2004 Ancient Lights Educational Media Published and Distributed by United Learning All rights to print materials cleared for classroom duplication and distribution. Name _________________ All About the Enlightenment: The Age of Reason 3 Video Quiz Answer each of the following questions either True or False: 1. -

Lesson 9: Coulomb's Law
Lesson 9: Coulomb's Law Charles Augustin de Coulomb Before getting into all the hardcore physics that surrounds him, it’s a good idea to understand a little about Coulomb. ● He was born in 1736 in Angoulême, France. ● He received the majority of his higher education at the Ecole du Genie at Mezieres (a french military university with a very high reputation, similar to universities like Oxford, Harvard, etc.) from which he graduated in 1761. ● He then spent some time serving as a military engineer in the West Indies and other French outposts, until 1781 when he was permanently stationed in Illustration 1: Paris and was able to devote more time to scientific research. Charles Coulomb Between 1785-91 he published seven memoirs (papers) on physics. ● One of them, published in 1785, discussed the inverse square law of forces between two charged particles. This just means that as you move charges apart, the force between them starts to decrease faster and faster (exponentially). ● In a later memoir he showed that the force is also proportional to the product of the charges, a relationship now called “Coulomb’s Law”. ● For his work, the unit of electrical charge is named after him. This is interesting in that Coulomb was one of the first people to help create the metric system. ● He died in 1806. The Torsion Balance When Coulomb was doing his original experiments he decided to use a torsion balance to measure the forces between charges. ● You already learned about a torsion balance in Physics 20 when you discussed Henry Cavendish’s experiment to measure the value of “G” , the universal gravitational constant. -

The French Revolution
The French Revolution People and Terms to know • Georges-Jacques Danton • Maximilien Robespierre • Jean-Paul Marat • Reign of Terror • Napoleon Bonaparte • National Convention • Universal Manhood Suffrage • Girondins • Jacobins • Committee of Public Safety • Revolutionary Tribunal • The Directory National Convention – senptember 1792 – 1st meeting Universal manhood suffrage – every man can vote Legislative Assembly – 3 main groups • Girodins o Dantonists – followed the lead of Georges Danton; believed Reign of Terror should end earlier and begin re-building of France • Conservatives • Jacobins o Radicals in Assembly; led by Maximilien Robespierre o Jacobins - divided- moderates/radicals § Robespierre § Jean-Paul Marat National Convention’s Actions • Declared end to monarchy • Beginning of new republic • Wrote new constitution • Brought Louis XVI to trial o Guilty – sentenced to death French Army – prior to Reign of Terror (Sept 1793) • Defeated Austrian and Prussian forces – stopped foreign invasion Opposition to the Constitution Arises • French people rose up against revolutionary government – “counterrevolutionary” o Ultimately wanted peace in France • Jacobins – Robespierre and Danton – Controlled National Convention • Arrested many Girodin delegates who opposed their policies • Charlotte Corday – Killed Jean-Paul Marat – arrested and executed Reign of Terror: suppress all opposition (Sept 1793- July 1794) • “annihilate internal and external enemies of the republic” • Executed Girondin opponents • Fought back European powers – gained -

Physical Implications of Coulomb's
INSTITUTE OF PHYSICS PUBLISHING METROLOGIA Metrologia 41 (2004) S159–S170 PII: S0026-1394(04)80313-X Physical implications of Coulomb’s Law G Spavieri1, G T Gillies2 and M Rodriguez3 1 Centro de Astrof´ısica Teorica,´ Facultad de Ciencias, Universidad de Los Andes, Merida,´ 5101 Venezuela 2 Department of Mechanical and Aerospace Engineering, University of Virginia, PO Box 400746, Charlottesville, VA 22904, USA 3 Departamento de F´ısica, FACYT, Universidad de Carabobo, Valencia, 2001 Venezuela E-mail: [email protected] and [email protected] Received 3 March 2004 Published 16 September 2004 Online at stacks.iop.org/Met/41/S159 doi:10.1088/0026-1394/41/5/S06 Abstract We examine the theoretical and experimental foundations of Coulomb’s Law and review the various roles it plays not only in electromagnetism and electrodynamics, but also in quantum mechanics, cosmology, and thermodynamics. The many implications of Coulomb’s Law draw attention to its fundamental importance within virtually all branches of physics and make this elementary yet profound law one of the most useful of all scientific tools. 1. Introductory historical outlook philosopher who had broad scientific interests in physics, electricity, magnetism, and optics, in addition to chemistry. He Few investigations in physics have enjoyed as sustained an was a politically involved Unitarian preacher and a sympathizer interest as have tests of Coulomb’s Law. As has been with the French Revolution, and these aspects of his life forced the case with most of the fundamental laws of physics, it him to move to America with his family in 1794. Priestley was discovered and elucidated through observations of basic is credited with the discovery of oxygen in 1774, which he phenomena. -

The Republic
SECTION 2 The Republic Getting Started As you BEFORE Y OU R EAD read, take 5SETHEInteractive Reader and Study Guide notes on the changes TOFAMILIARIZESTUDENTSWITHTHESECTION MAIN I DEA READING F OCUS KEY T ERMS AND P EOPLE made in French gov- CONTENT An extreme government 1. What changes did the radical Maximilien Robespierre ernment and society changed French society and government make in French guillotine and on the Reign of Interactive Reader and Study Guide, tried through harsh means society and politics? counterrevolution Terror. Section 2 to eliminate its critics within 2. What was the Reign of Reign of Terror France. Terror, and how did it end? The Republic Name _____________________________ Class _________________ Date __________________ The French Revolution and Napoleon Section 2 MAIN IDEA An extreme government changed French society and tried through harsh means to eliminate its critics within France. Key Terms and People Jean-Paul Marat Maximilien Robespierre Mountain member and a leader of the National Convention guillotine an execution device that drops a sharp, heavy blade through the victim’s neck counterrevolution a revolution against a government established by a revolution Reign of Terror series of accusations, arrests and executions started by the Mountain MUST DIE! Taking Notes As you read the summary, use a chart like the one below to record changes in French government and society as well as those brought about by the Reign of Terror. Jacques-Louis David painted the Death of Marat Original content Copyright © by Holt, Rinehart and Winston. Additions and changes to the original content are the responsibility of the instructor. -

Napoleon Bonaparte: His Successes and Failures
ISSN 2414-8385 (Online) European Journal of September-December 2017 ISSN 2414-8377 (Print Multidisciplinary Studies Volume 2, Issue 7 Napoleon Bonaparte: His Successes and Failures Zakia Sultana Assist. Prof., School of Liberal Arts and Social Sciences, University of Information Technology and Sciences (UITS), Baridhara, Dhaka, Bangladesh Abstract Napoleon Bonaparte (1769-1821), also known as Napoleon I, was a French military leader and emperor who conquered much of Europe in the early 19th century. Born on the island of Corsica, Napoleon rapidly rose through the ranks of the military during the French Revolution (1789-1799). After seizing political power in France in a 1799 coup d’état, he crowned himself emperor in 1804. Shrewd, ambitious and a skilled military strategist, Napoleon successfully waged war against various coalitions of European nations and expanded his empire. However, after a disastrous French invasion of Russia in 1812, Napoleon abdicated the throne two years later and was exiled to the island of Elba. In 1815, he briefly returned to power in his Hundred Days campaign. After a crushing defeat at the Battle of Waterloo, he abdicated once again and was exiled to the remote island of Saint Helena, where he died at 51.Napoleon was responsible for spreading the values of the French Revolution to other countries, especially in legal reform and the abolition of serfdom. After the fall of Napoleon, not only was the Napoleonic Code retained by conquered countries including the Netherlands, Belgium, parts of Italy and Germany, but has been used as the basis of certain parts of law outside Europe including the Dominican Republic, the US state of Louisiana and the Canadian province of Quebec.