Draft Genome of the Common Snapping Turtle, Chelydra Serpentina, a Model For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Xenosaurus Tzacualtipantecus. the Zacualtipán Knob-Scaled Lizard Is Endemic to the Sierra Madre Oriental of Eastern Mexico
Xenosaurus tzacualtipantecus. The Zacualtipán knob-scaled lizard is endemic to the Sierra Madre Oriental of eastern Mexico. This medium-large lizard (female holotype measures 188 mm in total length) is known only from the vicinity of the type locality in eastern Hidalgo, at an elevation of 1,900 m in pine-oak forest, and a nearby locality at 2,000 m in northern Veracruz (Woolrich- Piña and Smith 2012). Xenosaurus tzacualtipantecus is thought to belong to the northern clade of the genus, which also contains X. newmanorum and X. platyceps (Bhullar 2011). As with its congeners, X. tzacualtipantecus is an inhabitant of crevices in limestone rocks. This species consumes beetles and lepidopteran larvae and gives birth to living young. The habitat of this lizard in the vicinity of the type locality is being deforested, and people in nearby towns have created an open garbage dump in this area. We determined its EVS as 17, in the middle of the high vulnerability category (see text for explanation), and its status by the IUCN and SEMAR- NAT presently are undetermined. This newly described endemic species is one of nine known species in the monogeneric family Xenosauridae, which is endemic to northern Mesoamerica (Mexico from Tamaulipas to Chiapas and into the montane portions of Alta Verapaz, Guatemala). All but one of these nine species is endemic to Mexico. Photo by Christian Berriozabal-Islas. amphibian-reptile-conservation.org 01 June 2013 | Volume 7 | Number 1 | e61 Copyright: © 2013 Wilson et al. This is an open-access article distributed under the terms of the Creative Com- mons Attribution–NonCommercial–NoDerivs 3.0 Unported License, which permits unrestricted use for non-com- Amphibian & Reptile Conservation 7(1): 1–47. -

A Molecular Phylogeny for Marine Turtles: Trait Mapping, Rate
Proc. Natl. Acad. Sci. USA Vol. 90, pp. 5574-5577, June 1993 Evolution A molecular phylogeny for marine turtles: Trait mapping, rate assessment, and conservation relevance (conservation genetlcs/cytochrome b/molecular systematics/mitochondrial DNA) BRIAN W. BOWEN*, WILLIAM S. NELSON, AND JOHN C. AVISE Department of Genetics, University of Georgia, Athens, GA 30602 Contributed by John C. Avise, March 25, 1993 ABSTRACT Nucleotide sequences from the cytochrome b Table 1. A recent taxonomy for marine turtles, with problematic gene of mitochondrial DNA were employed to resolve phylo- areas indicated by parenthetical questions genetic controversies and to assess molecular evolutionary rates Order Testudines-all extant turtles, freshwater, terrestrial, and in marine turtles (Chelonioidea). Findings of special relevance marine to conservation biology include discovery of a distant relation- Family Dermochelyidae-Dermochelys coriacea (leatherback) ship betweenNatatorand other chelonild species, the paraphyly (Are marine turtles monophyletic? Where does this species lie ofChelonia mydas with respect to Chelonia agassizi, and genetic with regard to broader turtle phylogeny?) distinctiveness of Lepidochelys kempi from Lepidochelys oliva- Family Cheloniidae cea. A longstanding debate in evolutionary ecology was re- Tribe Chelonini solved by phylogenetic mapping of dietary habits, which indi- Genus Chelonia-C. mydas (green) cates that the spongivore Ereinochelys imbricata evolved from C. agassizi (black) a carnivorous rather than a herbivorous ancestor. Sequence (Are these two forms distinct species?) divergences at intergeneric and interfamilial levels, when as- Genus Natator-N. depressus (flatback) sessed against fossil-based separation times, support previous (Is this species a close ally of the green turtle, or perhaps suggestions (from microevolutionary comparisons) that mito- allied more closely to members of the Carettini?) chondrial DNA in marine turtles evolves much more slowly Tribe Carettini than under the "conventional" vertebrate clock. -
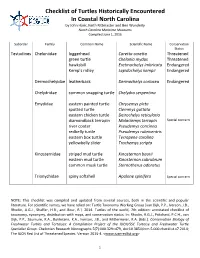
N.C. Turtles Checklist
Checklist of Turtles Historically Encountered In Coastal North Carolina by John Hairr, Keith Rittmaster and Ben Wunderly North Carolina Maritime Museums Compiled June 1, 2016 Suborder Family Common Name Scientific Name Conservation Status Testudines Cheloniidae loggerhead Caretta caretta Threatened green turtle Chelonia mydas Threatened hawksbill Eretmochelys imbricata Endangered Kemp’s ridley Lepidochelys kempii Endangered Dermochelyidae leatherback Dermochelys coriacea Endangered Chelydridae common snapping turtle Chelydra serpentina Emydidae eastern painted turtle Chrysemys picta spotted turtle Clemmys guttata eastern chicken turtle Deirochelys reticularia diamondback terrapin Malaclemys terrapin Special concern river cooter Pseudemys concinna redbelly turtle Pseudemys rubriventris eastern box turtle Terrapene carolina yellowbelly slider Trachemys scripta Kinosternidae striped mud turtle Kinosternon baurii eastern mud turtle Kinosternon subrubrum common musk turtle Sternotherus odoratus Trionychidae spiny softshell Apalone spinifera Special concern NOTE: This checklist was compiled and updated from several sources, both in the scientific and popular literature. For scientific names, we have relied on: Turtle Taxonomy Working Group [van Dijk, P.P., Iverson, J.B., Rhodin, A.G.J., Shaffer, H.B., and Bour, R.]. 2014. Turtles of the world, 7th edition: annotated checklist of taxonomy, synonymy, distribution with maps, and conservation status. In: Rhodin, A.G.J., Pritchard, P.C.H., van Dijk, P.P., Saumure, R.A., Buhlmann, K.A., Iverson, J.B., and Mittermeier, R.A. (Eds.). Conservation Biology of Freshwater Turtles and Tortoises: A Compilation Project of the IUCN/SSC Tortoise and Freshwater Turtle Specialist Group. Chelonian Research Monographs 5(7):000.329–479, doi:10.3854/crm.5.000.checklist.v7.2014; The IUCN Red List of Threatened Species. -

Gopher Tortoise Demographic Responses to a Novel Disturbance Regime
The Journal of Wildlife Management 1–10; 2019; DOI: 10.1002/jwmg.21774 Research Article Gopher Tortoise Demographic Responses to a Novel Disturbance Regime HUNTER J. HOWELL ,1,2 Department of Biology, University of Miami, 1301 Memorial Drive, Coral Gables, FL 33146, USA BETSIE B. ROTHERMEL,2 Archbold Biological Station, 123 Main Drive, Venus, FL 33960, USA K. NICOLE WHITE, Daniel B. Warnell School of Forestry and Natural Resources, University of Georgia, 180 E Green Street, Athens, GA 30602, USA CHRISTOPHER A. SEARCY, Department of Biology, University of Miami, 1301 Memorial Drive, Coral Gables, FL 33146, USA ABSTRACT The long‐term viability of gopher tortoise (Gopherus polyphemus) populations is jeopardized by increased urbanization and habitat degradation owing to fire suppression. Because the species’ remaining natural habitats in the southeastern United States exist within a mosaic of anthropogenic land uses, it is important to understand demographic responses to contrasting land uses and habitat management regimes. We examined differences in demographic parameters among fire‐suppressed sandhill, restored sandhill, and former sandhill (i.e., ruderal) land use‐land cover (LULC) types at Archbold Biological Station in south‐ central Florida, USA. Using Program MARK, we estimated population size, and sex‐specific and LULC‐specific survivorship based on 6 years of mark‐recapture data. We also analyzed individual growth trajectories and clutch sizes to determine whether growth rates or reproductive output differed among LULC types. Tortoises in an open, ruderal field occurred at a higher density (7.79/ha) than in adjacent restored (1.43/ha) or fire‐suppressed (0.40/ha) sandhill. Despite this higher density, both adult survivorship and body size were significantly higher in the ruderal field. -

The Conservation Biology of Tortoises
The Conservation Biology of Tortoises Edited by Ian R. Swingland and Michael W. Klemens IUCN/SSC Tortoise and Freshwater Turtle Specialist Group and The Durrell Institute of Conservation and Ecology Occasional Papers of the IUCN Species Survival Commission (SSC) No. 5 IUCN—The World Conservation Union IUCN Species Survival Commission Role of the SSC 3. To cooperate with the World Conservation Monitoring Centre (WCMC) The Species Survival Commission (SSC) is IUCN's primary source of the in developing and evaluating a data base on the status of and trade in wild scientific and technical information required for the maintenance of biological flora and fauna, and to provide policy guidance to WCMC. diversity through the conservation of endangered and vulnerable species of 4. To provide advice, information, and expertise to the Secretariat of the fauna and flora, whilst recommending and promoting measures for their con- Convention on International Trade in Endangered Species of Wild Fauna servation, and for the management of other species of conservation concern. and Flora (CITES) and other international agreements affecting conser- Its objective is to mobilize action to prevent the extinction of species, sub- vation of species or biological diversity. species, and discrete populations of fauna and flora, thereby not only maintain- 5. To carry out specific tasks on behalf of the Union, including: ing biological diversity but improving the status of endangered and vulnerable species. • coordination of a programme of activities for the conservation of biological diversity within the framework of the IUCN Conserva- tion Programme. Objectives of the SSC • promotion of the maintenance of biological diversity by monitor- 1. -

Relative Abundance, Population Structure, and Habitat
University of Missouri, St. Louis IRL @ UMSL Theses Graduate Works 11-18-2010 Relative Abundance, Population Structure, and Habitat Utilization of the Alligator Snapping Turtle (Macrochelys temminckii) and Eastern Snapping Turtle (Chelydra serpentina) in southeastern Missouri Timothy Charles Lescher University of Missouri-St. Louis, [email protected] Follow this and additional works at: http://irl.umsl.edu/thesis Recommended Citation Lescher, Timothy Charles, "Relative Abundance, Population Structure, and Habitat Utilization of the Alligator Snapping Turtle (Macrochelys temminckii) and Eastern Snapping Turtle (Chelydra serpentina) in southeastern Missouri" (2010). Theses. 44. http://irl.umsl.edu/thesis/44 This Thesis is brought to you for free and open access by the Graduate Works at IRL @ UMSL. It has been accepted for inclusion in Theses by an authorized administrator of IRL @ UMSL. For more information, please contact [email protected]. Relative Abundance, Population Structure, and Habitat Utilization of the Alligator Snapping Turtle (Macrochelys temminckii) and Eastern Snapping Turtle (Chelydra serpentina) in southeastern Missouri Timothy C. Lescher B.S., Biology, University of Missouri-Columbia, 2001 A Thesis Submitted to the Graduate School at the University of Missouri-Saint Louis in partial fulfillment of the requirements for the degree Master’s of Science in Biology November 2010 Advisory Committee Zuleyma Tang-Martinez, Ph.D. Chair Jeffrey T. Briggler, Ph.D. Bette A. Loiselle, Ph.D. Copyright, Timothy C. Lescher, 2010 1 TABLE -

The Common Snapping Turtle, Chelydra Serpentina
The Common Snapping Turtle, Chelydra serpentina Rylen Nakama FISH 423: Olden 12/5/14 Figure 1. The Common Snapping Turtle, one of the most widespread reptiles in North America. Photo taken in Quebec, Canada. Image from https://www.flickr.com/photos/yorthopia/7626614760/. Classification Order: Testudines Family: Chelydridae Genus: Chelydra Species: serpentina (Linnaeus, 1758) Previous research on Chelydra serpentina (Phillips et al., 1996) acknowledged four subspecies, C. s. serpentina (Northern U.S. and Figure 2. Side profile of Chelydra serpentina. Note Canada), C. s. osceola (Southeastern U.S.), C. s. the serrated posterior end of the carapace and the rossignonii (Central America), and C. s. tail’s raised central ridge. Photo from http://pelotes.jea.com/AnimalFact/Reptile/snapturt.ht acutirostris (South America). Recent IUCN m. reclassification of chelonians based on genetic analyses (Rhodin et al., 2010) elevated C. s. rossignonii and C. s. acutirostris to species level and established C. s. osceola as a synonym for C. s. serpentina, thus eliminating subspecies within C. serpentina. Antiquated distinctions between the two formerly recognized North American subspecies were based on negligible morphometric variations between the two populations. Interbreeding in the overlapping range of the two populations was well documented, further discrediting the validity of the subspecies distinction (Feuer, 1971; Aresco and Gunzburger, 2007). Therefore, any emphasis of subspecies differentiation in the ensuing literature should be disregarded. Figure 3. Front-view of a captured Chelydra Continued usage of invalid subspecies names is serpentina. Different skin textures and the distinctive pink mouth are visible from this angle. Photo from still prevalent in the exotic pet trade for C. -

UC Davis UC Davis Previously Published Works
UC Davis UC Davis Previously Published Works Title Survival, Demography, and Growth of Gopher Tortoises (Gopherus polyphemus) from Three Study Sites with Different Management Histories Permalink https://escholarship.org/uc/item/9sc747h0 Journal JOURNAL OF WILDLIFE MANAGEMENT, 78(7) ISSN 0022-541X Authors Tuberville, Tracey D Todd, Brian D Hermann, Sharon M et al. Publication Date 2014-09-01 DOI 10.1002/jwmg.773 Peer reviewed eScholarship.org Powered by the California Digital Library University of California The Journal of Wildlife Management 78(7):1151–1160; 2014; DOI: 10.1002/jwmg.773 Research Article Survival, Demography, and Growth of Gopher Tortoises (Gopherus polyphemus) from Three Study Sites with Different Management Histories TRACEY D. TUBERVILLE,1 University of Georgia’s Savannah River Ecology Lab, Drawer E, Aiken, SC 29802, USA BRIAN D. TODD, Department of Wildlife, Fish, and Conservation Biology, University of California, Davis, One Shields Ave, Davis, CA 95616, USA SHARON M. HERMANN, Department of Biological Sciences, Auburn University, Auburn, AL 36849, USA WILLIAM K. MICHENER, University Libraries, 1312 Basehart SE, Albuquerque, NM 87106, USA CRAIG GUYER, Department of Biological Sciences, Auburn University, Auburn, AL 36849, USA ABSTRACT Recovery or sustainable management of wildlife populations often entails management of habitat on which they depend. In this regard, turtles pose unique conservation challenges because of their life histories. The combination of late maturity, low survival when young, and dependence on high adult survival suggests they may be slow to respond demographically to conventional habitat management. Thus, long- term studies are necessary to understand population dynamics and recovery potential in these species. -
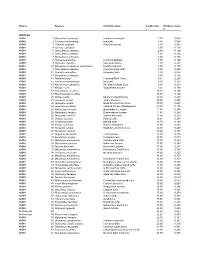
PDF File Containing Table of Lengths and Thicknesses of Turtle Shells And
Source Species Common name length (cm) thickness (cm) L t TURTLES AMNH 1 Sternotherus odoratus common musk turtle 2.30 0.089 AMNH 2 Clemmys muhlenbergi bug turtle 3.80 0.069 AMNH 3 Chersina angulata Angulate tortoise 3.90 0.050 AMNH 4 Testudo carbonera 6.97 0.130 AMNH 5 Sternotherus oderatus 6.99 0.160 AMNH 6 Sternotherus oderatus 7.00 0.165 AMNH 7 Sternotherus oderatus 7.00 0.165 AMNH 8 Homopus areolatus Common padloper 7.95 0.100 AMNH 9 Homopus signatus Speckled tortoise 7.98 0.231 AMNH 10 Kinosternon subrabum steinochneri Florida mud turtle 8.90 0.178 AMNH 11 Sternotherus oderatus Common musk turtle 8.98 0.290 AMNH 12 Chelydra serpentina Snapping turtle 8.98 0.076 AMNH 13 Sternotherus oderatus 9.00 0.168 AMNH 14 Hardella thurgi Crowned River Turtle 9.04 0.263 AMNH 15 Clemmys muhlenbergii Bog turtle 9.09 0.231 AMNH 16 Kinosternon subrubrum The Eastern Mud Turtle 9.10 0.253 AMNH 17 Kinixys crosa hinged-back tortoise 9.34 0.160 AMNH 18 Peamobates oculifers 10.17 0.140 AMNH 19 Peammobates oculifera 10.27 0.140 AMNH 20 Kinixys spekii Speke's hinged tortoise 10.30 0.201 AMNH 21 Terrapene ornata ornate box turtle 10.30 0.406 AMNH 22 Terrapene ornata North American box turtle 10.76 0.257 AMNH 23 Geochelone radiata radiated tortoise (Madagascar) 10.80 0.155 AMNH 24 Malaclemys terrapin diamondback terrapin 11.40 0.295 AMNH 25 Malaclemys terrapin Diamondback terrapin 11.58 0.264 AMNH 26 Terrapene carolina eastern box turtle 11.80 0.259 AMNH 27 Chrysemys picta Painted turtle 12.21 0.267 AMNH 28 Chrysemys picta painted turtle 12.70 0.168 AMNH 29 -

Snapping Turtle, Chelydra Serpentina, Overland Movements Near the Southeastern Extent of Its Range David A
Georgia Journal of Science Volume 68 No. 2 Scholarly Contributions from the Article 11 Membership and Others 2010 Snapping Turtle, Chelydra serpentina, Overland Movements Near the Southeastern Extent of its Range David A. Steen [email protected] Sean C. Sterrett Aubrey M. Heupel Lora L. Smith Follow this and additional works at: https://digitalcommons.gaacademy.org/gjs Part of the Life Sciences Commons Recommended Citation Steen, David A.; Sterrett, Sean C.; Heupel, Aubrey M.; and Smith, Lora L. (2010) "Snapping Turtle, Chelydra serpentina, Overland Movements Near the Southeastern Extent of its Range," Georgia Journal of Science, Vol. 68, No. 2, Article 11. Available at: https://digitalcommons.gaacademy.org/gjs/vol68/iss2/11 This Research Articles is brought to you for free and open access by Digital Commons @ the Georgia Academy of Science. It has been accepted for inclusion in Georgia Journal of Science by an authorized editor of Digital Commons @ the Georgia Academy of Science. 196 Steen et al.: Snapping Turtle Overland Movements SNAPPING Turtle, CHELYDRA SERPENTINA, OVERLAND MOVEMENTS NEAR THE SOUTHEASTERN EXTENT OF ITS RANGE David A. Steen1,2*, Sean C. Sterrett2, Aubrey M. Heupel2 and Lora L. Smith2 1Auburn University, 331 Funchess Hall, Auburn, Alabama, 36849 2Joseph W. Jones Ecological Research Center Route 2, Box 2324, Newton, GA 39870 Institution at which work was completed: Joseph W. Jones Ecological Research Center * Corresponding author: [email protected] ABSTRACT Terrestrial movements of turtles are of interest due to the conserva- tion implications for this imperiled group and the general lack of information on this topic, particularly in wide-ranging species. -

The PARI Journal Vol. VI, No. 3
ThePARIJournal A quarterly publication of the Pre-Columbian Art Research Institute Volume VI, No.3, Winter 2006 Teasing the Turtle from its Shell: In This Issue: AHKand MAHK in MayaWriting* Teasing the Turtle MARC ZENDER from its Shell: Peabody Museum of Archaeology & Ethnology, Harvard University AHK and MAHK in Maya Writing resents the profile carapace of a testudine by Marc Zender (i.e., a turtle, tortoise or terrapin), careful PAGES 1-14 examination of their formal features, dis- tributions and lexical associations reveals • them to have been quite distinct entities, Morley's Diary: rigidly distinguished by scribes through- a b out the life of the script. As discussed in April 23, 1932 detail below, there is in fact strong reason Figure 1. “Turtle Shell” Glyphs: (a) MAHK “carapace” and PAGES 15-16 “cover”; (b) AHK “turtle.” All drawings by the author. to see the first sign (Figure 1a) as the de- piction of a turtle shell carrying the value Formally similar but nevertheless dis- MAHK “carapace, shell,” and, less specifi- tinct signs remain a constant challenge to cally, “cover, enclosure.” By contrast, the Joel Skidmore students of the ancient Maya script. Pre- latter sign (Figure 1b), while also depict- Editor ing a turtle shell, has long been known [email protected] viously I have argued that a number of signs traditionally considered identical to carry the value AHK “turtle,” and is are in fact quite distinct, as demonstrated therefore best thought of as the pars pro The PARI Journal by formal divergences and consistent dis- toto depiction of a “turtle” proper. -

Terrapene Carolina (Linnaeus 1758) – Eastern Box Turtle, Common Box Turtle
Conservation Biology of Freshwater Turtles and Tortoises: A Compilation Project ofEmydidae the IUCN/SSC — TortoiseTerrapene and Freshwatercarolina Turtle Specialist Group 085.1 A.G.J. Rhodin, P.C.H. Pritchard, P.P. van Dijk, R.A. Saumure, K.A. Buhlmann, J.B. Iverson, and R.A. Mittermeier, Eds. Chelonian Research Monographs (ISSN 1088-7105) No. 5, doi:10.3854/crm.5.085.carolina.v1.2015 © 2015 by Chelonian Research Foundation • Published 26 January 2015 Terrapene carolina (Linnaeus 1758) – Eastern Box Turtle, Common Box Turtle A. ROSS KIESTER1 AND LISABETH L. WILLEY2 1Turtle Conservancy, 49 Bleecker St., Suite 601, New York, New York 10012 USA [[email protected]]; 2Department of Environmental Studies, Antioch University New England, 40 Avon St., Keene, New Hampshire 03431 USA [[email protected]] SUMMARY. – The Eastern Box Turtle, Terrapene carolina (Family Emydidae), as currently understood, contains six living subspecies of small turtles (carapace lengths to ca. 115–235 mm) able to close their hinged plastrons into a tightly closed box. Although the nominate subspecies is among the most widely distributed and well-known of the world’s turtles, the two Mexican subspecies are poorly known. This primarily terrestrial, though occasionally semi-terrestrial, species ranges throughout the eastern and southern United States and disjunctly in Mexico. It was generally recognized as common in the USA throughout the 20th century, but is now threatened by continuing habitat conversion, road mortality, and collection for the pet trade, and notable population declines have been documented throughout its range. In the United States, this turtle is a paradigm example of the conservation threats that beset and impact a historically common North American species.