Marine Turtle Mitogenome Phylogenetics and Evolution
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Beach Dynamics and Impact of Armouring on Olive Ridley Sea Turtle (Lepidochelys Olivacea) Nesting at Gahirmatha Rookery of Odisha Coast, India
Indian Journal of Geo-Marine Sciences Vol. 45(2), February 2016, pp. 233-238 Beach dynamics and impact of armouring on olive ridley sea turtle (Lepidochelys olivacea) nesting at Gahirmatha rookery of Odisha coast, India Satyaranjan Behera1, 2, Basudev Tripathy3*, K. Sivakumar2, B.C. Choudhury2 1Odisha Biodiversity Board, Regional Plant Resource Centre Campus, Nayapalli, Bhubaneswar-15 2Wildlife Institute of India, Dehradun, PO Box 18, Chandrabani, Dehradun – 248 001, India. 3Zoological Survey of India, Prani Vigyan Bhawan, M-Block, New Alipore, Kolkata-700 053 (India) *[E. mail:[email protected]] Received 28 March 2014; revised 18 September 2014 Gahirmatha arribada beach are most dynamic and eroding at a faster rate over the years from 2008-09 to 2010-11, especially during the turtles breeding seasons. Impact of armouring cement tetrapod on olive ridley sea turtle nesting beach at Gahirmatha rookery of Odisha coast has also been reported in this study. This study documented the area of nesting beach has reduced from 0.07 km2to 0.06 km2. Due to a constraint of nesting space, turtles were forced to nest in the gap of cement tetrapods adjacent to the arribada beach and get entangled there, resulting into either injury or death. A total of 209 and 24 turtles were reported to be injured and dead due to placement of cement tetrapods in their nesting beach during 2008-09 and 2010-11 respectively. Olive ridley turtles in Odisha are now exposed to many problems other than fishing related casualty and precautionary measures need to be taken by the wildlife and forest authorities to safeguard the Olive ridleys and their nesting habitat at Gahirmatha. -
Fly High Dive Deep
FLY HIGH DIVE DEEP COMMERCIAL DIVING REMOTE OPERATED VEHICLES SPACE EXPLORATION HUMAN LIFE SCIENCE WWW.BLUEABYSS.UK THE PROMISE Blue Abyss is among the most ground-breaking projects of its time. Designed to support the commercial diving, remote operated vehicle, human spaceflight and human life science sectors, Blue Abyss promises to be Europe’s premier extreme environment research, development and training facility. This unique aquatic centre will house the world’s largest and deepest indoor pool, alongside: hyper and hypobaric chambers; the Kuehnegger Human Performance Centre; a micro-gravity simulation suspension suite for replicating the effects of weightlessness and hypo-gravity; amphitheatre and classrooms; cafeteria and 120-bed hotel. ASTRONAUTS AND “OTHER SPACE PROFESSIONALS WILL WANT TO COME FROM AROUND THE WORLD TO USE THE MASSIVE, YET CONTROLLED, ENVIRONMENT TO REDUCE RISK IN SPACE. I CAN SEE PLENTY OF INTERNATIONAL COLLABORATIONS AND BUSINESS VENTURES STARTING LIFE WITHIN BLUE ABYSS. DR HELEN SHARMAN” FIRST BRITISH CITIZEN IN SPACE 1 Full onsite mission control, hypo and hyperbaric chambers Crane and lifting platform (30 tonnes) Training/experience mock-ups Pool 50m x 40m on surface Multi-level functionality including ‘Astrolab’ at 12m 50m at deepest point / THE MULTI-LEVEL POOL WILL CONTAIN 38,000M3 OF WATER, EXCEEDING ALL OTHER FACILITIES IN EXISTENCE BOTH IN TERMS OF VOLUME AND DEPTH. Image courtesy of Cityscape Digital 2 3 THE POSSIBILITIES Blue Abyss is a truly pioneering project that will extend the Blue Abyss is designed to cater for Space environment simulation possibilities for education, commercial and scientific research, on one hand, and freediving on the other, with a huge variety of development and training beyond anything that exists today. -

A Molecular Phylogeny for Marine Turtles: Trait Mapping, Rate
Proc. Natl. Acad. Sci. USA Vol. 90, pp. 5574-5577, June 1993 Evolution A molecular phylogeny for marine turtles: Trait mapping, rate assessment, and conservation relevance (conservation genetlcs/cytochrome b/molecular systematics/mitochondrial DNA) BRIAN W. BOWEN*, WILLIAM S. NELSON, AND JOHN C. AVISE Department of Genetics, University of Georgia, Athens, GA 30602 Contributed by John C. Avise, March 25, 1993 ABSTRACT Nucleotide sequences from the cytochrome b Table 1. A recent taxonomy for marine turtles, with problematic gene of mitochondrial DNA were employed to resolve phylo- areas indicated by parenthetical questions genetic controversies and to assess molecular evolutionary rates Order Testudines-all extant turtles, freshwater, terrestrial, and in marine turtles (Chelonioidea). Findings of special relevance marine to conservation biology include discovery of a distant relation- Family Dermochelyidae-Dermochelys coriacea (leatherback) ship betweenNatatorand other chelonild species, the paraphyly (Are marine turtles monophyletic? Where does this species lie ofChelonia mydas with respect to Chelonia agassizi, and genetic with regard to broader turtle phylogeny?) distinctiveness of Lepidochelys kempi from Lepidochelys oliva- Family Cheloniidae cea. A longstanding debate in evolutionary ecology was re- Tribe Chelonini solved by phylogenetic mapping of dietary habits, which indi- Genus Chelonia-C. mydas (green) cates that the spongivore Ereinochelys imbricata evolved from C. agassizi (black) a carnivorous rather than a herbivorous ancestor. Sequence (Are these two forms distinct species?) divergences at intergeneric and interfamilial levels, when as- Genus Natator-N. depressus (flatback) sessed against fossil-based separation times, support previous (Is this species a close ally of the green turtle, or perhaps suggestions (from microevolutionary comparisons) that mito- allied more closely to members of the Carettini?) chondrial DNA in marine turtles evolves much more slowly Tribe Carettini than under the "conventional" vertebrate clock. -

Monofins for Freediving
Monofins for Freediving We have been intermittently following the debate concerning the use of the monofin in freediving and would like to share some of our findings. Two years ago we put together the first experimental monofin/freedive clinic where we assembled some unique elements. We put together the leading trainers in monofin swimming, namely the Russian coaches from Tomsk university, who train both the Russian national team and their chief rivals, the Chinese, the leading specialist monofin manufacturer belonging to the same school and a group of freedivers which represented the best cross-section, from the very top of freediving competition to the very novice. This same group also represented advanced freedivers who already had experience with the monofin, advanced freedivers who had never used a monofin and a novice freediver with no experience of the monofin. Although the number of freedivers involved was small we feel that with a larger group the conclusions would have been much the same. The objectives were to find (i) What style and why? (ii) What rhythm and amplitude of movement? (iii) What kind of monofin and what stiffness of blade and if this was individual what the relevant criteria for monofin choice should be? (iv) What compromises and adaptations had to be made to suit the specific needs of the freediver? (v) What was the best training method for the monofin freediver. What style and why? We had heard a lot of talk concerning adaptations of the ‘classic ’style that freedivers should adopt. I know from personal acquaintance that some of the people recommending various adaptations were not capable of demonstrating a good classic style hence their recommendations were from lack of ability in the monofin and hence lack of choice through limited ability. -

AN INTRODUCTION to Texas Turtles
TEXAS PARKS AND WILDLIFE AN INTRODUCTION TO Texas Turtles Mark Klym An Introduction to Texas Turtles Turtle, tortoise or terrapin? Many people get confused by these terms, often using them interchangeably. Texas has a single species of tortoise, the Texas tortoise (Gopherus berlanderi) and a single species of terrapin, the diamondback terrapin (Malaclemys terrapin). All of the remaining 28 species of the order Testudines found in Texas are called “turtles,” although some like the box turtles (Terrapene spp.) are highly terrestrial others are found only in marine (saltwater) settings. In some countries such as Great Britain or Australia, these terms are very specific and relate to the habit or habitat of the animal; in North America they are denoted using these definitions. Turtle: an aquatic or semi-aquatic animal with webbed feet. Tortoise: a terrestrial animal with clubbed feet, domed shell and generally inhabiting warmer regions. Whatever we call them, these animals are a unique tie to a period of earth’s history all but lost in the living world. Turtles are some of the oldest reptilian species on the earth, virtually unchanged in 200 million years or more! These slow-moving, tooth less, egg-laying creatures date back to the dinosaurs and still retain traits they used An Introduction to Texas Turtles | 1 to survive then. Although many turtles spend most of their lives in water, they are air-breathing animals and must come to the surface to breathe. If they spend all this time in water, why do we see them on logs, rocks and the shoreline so often? Unlike birds and mammals, turtles are ectothermic, or cold- blooded, meaning they rely on the temperature around them to regulate their body temperature. -

Background on Sea Turtles
Background on Sea Turtles Five of the seven species of sea turtles call Virginia waters home between the months of April and November. All five species are listed on the U.S. List of Endangered and Threatened Wildlife and Plants and classified as either “Threatened” or “Endangered”. It is estimated that anywhere between five and ten thousand sea turtles enter the Chesapeake Bay during the spring and summer months. Of these the most common visitor is the loggerhead followed by the Kemp’s ridley, leatherback and green. The least common of the five species is the hawksbill. The Loggerhead is the largest hard-shelled sea turtle often reaching weights of 1000 lbs. However, the ones typically sighted in Virginia’s waters range in size from 50 to 300 lbs. The diet of the loggerhead is extensive including jellies, sponges, bivalves, gastropods, squid and shrimp. While visiting the Bay waters the loggerhead dines almost exclusively on horseshoe crabs. Virginia is the northern most nesting grounds for the loggerhead. Because the temperature of the nest dictates the sex of the turtle it is often thought that the few nests found in Virginia are producing predominately male offspring. Once the male turtles enter the water they will never return to land in their lifetime. Loggerheads are listed as a “Threatened” species. The Kemp’s ridley sea turtle is the second most frequent visitor in Virginia waters. It is the smallest of the species off Virginia’s coast reaching a maximum weight of just over 100 lbs. The specimens sited in Virginia are often less than 30 lbs. -
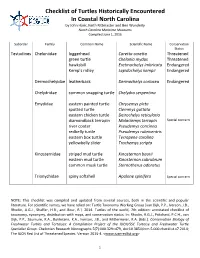
N.C. Turtles Checklist
Checklist of Turtles Historically Encountered In Coastal North Carolina by John Hairr, Keith Rittmaster and Ben Wunderly North Carolina Maritime Museums Compiled June 1, 2016 Suborder Family Common Name Scientific Name Conservation Status Testudines Cheloniidae loggerhead Caretta caretta Threatened green turtle Chelonia mydas Threatened hawksbill Eretmochelys imbricata Endangered Kemp’s ridley Lepidochelys kempii Endangered Dermochelyidae leatherback Dermochelys coriacea Endangered Chelydridae common snapping turtle Chelydra serpentina Emydidae eastern painted turtle Chrysemys picta spotted turtle Clemmys guttata eastern chicken turtle Deirochelys reticularia diamondback terrapin Malaclemys terrapin Special concern river cooter Pseudemys concinna redbelly turtle Pseudemys rubriventris eastern box turtle Terrapene carolina yellowbelly slider Trachemys scripta Kinosternidae striped mud turtle Kinosternon baurii eastern mud turtle Kinosternon subrubrum common musk turtle Sternotherus odoratus Trionychidae spiny softshell Apalone spinifera Special concern NOTE: This checklist was compiled and updated from several sources, both in the scientific and popular literature. For scientific names, we have relied on: Turtle Taxonomy Working Group [van Dijk, P.P., Iverson, J.B., Rhodin, A.G.J., Shaffer, H.B., and Bour, R.]. 2014. Turtles of the world, 7th edition: annotated checklist of taxonomy, synonymy, distribution with maps, and conservation status. In: Rhodin, A.G.J., Pritchard, P.C.H., van Dijk, P.P., Saumure, R.A., Buhlmann, K.A., Iverson, J.B., and Mittermeier, R.A. (Eds.). Conservation Biology of Freshwater Turtles and Tortoises: A Compilation Project of the IUCN/SSC Tortoise and Freshwater Turtle Specialist Group. Chelonian Research Monographs 5(7):000.329–479, doi:10.3854/crm.5.000.checklist.v7.2014; The IUCN Red List of Threatened Species. -

Gopher Tortoise Demographic Responses to a Novel Disturbance Regime
The Journal of Wildlife Management 1–10; 2019; DOI: 10.1002/jwmg.21774 Research Article Gopher Tortoise Demographic Responses to a Novel Disturbance Regime HUNTER J. HOWELL ,1,2 Department of Biology, University of Miami, 1301 Memorial Drive, Coral Gables, FL 33146, USA BETSIE B. ROTHERMEL,2 Archbold Biological Station, 123 Main Drive, Venus, FL 33960, USA K. NICOLE WHITE, Daniel B. Warnell School of Forestry and Natural Resources, University of Georgia, 180 E Green Street, Athens, GA 30602, USA CHRISTOPHER A. SEARCY, Department of Biology, University of Miami, 1301 Memorial Drive, Coral Gables, FL 33146, USA ABSTRACT The long‐term viability of gopher tortoise (Gopherus polyphemus) populations is jeopardized by increased urbanization and habitat degradation owing to fire suppression. Because the species’ remaining natural habitats in the southeastern United States exist within a mosaic of anthropogenic land uses, it is important to understand demographic responses to contrasting land uses and habitat management regimes. We examined differences in demographic parameters among fire‐suppressed sandhill, restored sandhill, and former sandhill (i.e., ruderal) land use‐land cover (LULC) types at Archbold Biological Station in south‐ central Florida, USA. Using Program MARK, we estimated population size, and sex‐specific and LULC‐specific survivorship based on 6 years of mark‐recapture data. We also analyzed individual growth trajectories and clutch sizes to determine whether growth rates or reproductive output differed among LULC types. Tortoises in an open, ruderal field occurred at a higher density (7.79/ha) than in adjacent restored (1.43/ha) or fire‐suppressed (0.40/ha) sandhill. Despite this higher density, both adult survivorship and body size were significantly higher in the ruderal field. -

The Conservation Biology of Tortoises
The Conservation Biology of Tortoises Edited by Ian R. Swingland and Michael W. Klemens IUCN/SSC Tortoise and Freshwater Turtle Specialist Group and The Durrell Institute of Conservation and Ecology Occasional Papers of the IUCN Species Survival Commission (SSC) No. 5 IUCN—The World Conservation Union IUCN Species Survival Commission Role of the SSC 3. To cooperate with the World Conservation Monitoring Centre (WCMC) The Species Survival Commission (SSC) is IUCN's primary source of the in developing and evaluating a data base on the status of and trade in wild scientific and technical information required for the maintenance of biological flora and fauna, and to provide policy guidance to WCMC. diversity through the conservation of endangered and vulnerable species of 4. To provide advice, information, and expertise to the Secretariat of the fauna and flora, whilst recommending and promoting measures for their con- Convention on International Trade in Endangered Species of Wild Fauna servation, and for the management of other species of conservation concern. and Flora (CITES) and other international agreements affecting conser- Its objective is to mobilize action to prevent the extinction of species, sub- vation of species or biological diversity. species, and discrete populations of fauna and flora, thereby not only maintain- 5. To carry out specific tasks on behalf of the Union, including: ing biological diversity but improving the status of endangered and vulnerable species. • coordination of a programme of activities for the conservation of biological diversity within the framework of the IUCN Conserva- tion Programme. Objectives of the SSC • promotion of the maintenance of biological diversity by monitor- 1. -

Relative Abundance, Population Structure, and Habitat
University of Missouri, St. Louis IRL @ UMSL Theses Graduate Works 11-18-2010 Relative Abundance, Population Structure, and Habitat Utilization of the Alligator Snapping Turtle (Macrochelys temminckii) and Eastern Snapping Turtle (Chelydra serpentina) in southeastern Missouri Timothy Charles Lescher University of Missouri-St. Louis, [email protected] Follow this and additional works at: http://irl.umsl.edu/thesis Recommended Citation Lescher, Timothy Charles, "Relative Abundance, Population Structure, and Habitat Utilization of the Alligator Snapping Turtle (Macrochelys temminckii) and Eastern Snapping Turtle (Chelydra serpentina) in southeastern Missouri" (2010). Theses. 44. http://irl.umsl.edu/thesis/44 This Thesis is brought to you for free and open access by the Graduate Works at IRL @ UMSL. It has been accepted for inclusion in Theses by an authorized administrator of IRL @ UMSL. For more information, please contact [email protected]. Relative Abundance, Population Structure, and Habitat Utilization of the Alligator Snapping Turtle (Macrochelys temminckii) and Eastern Snapping Turtle (Chelydra serpentina) in southeastern Missouri Timothy C. Lescher B.S., Biology, University of Missouri-Columbia, 2001 A Thesis Submitted to the Graduate School at the University of Missouri-Saint Louis in partial fulfillment of the requirements for the degree Master’s of Science in Biology November 2010 Advisory Committee Zuleyma Tang-Martinez, Ph.D. Chair Jeffrey T. Briggler, Ph.D. Bette A. Loiselle, Ph.D. Copyright, Timothy C. Lescher, 2010 1 TABLE -

The Common Snapping Turtle, Chelydra Serpentina
The Common Snapping Turtle, Chelydra serpentina Rylen Nakama FISH 423: Olden 12/5/14 Figure 1. The Common Snapping Turtle, one of the most widespread reptiles in North America. Photo taken in Quebec, Canada. Image from https://www.flickr.com/photos/yorthopia/7626614760/. Classification Order: Testudines Family: Chelydridae Genus: Chelydra Species: serpentina (Linnaeus, 1758) Previous research on Chelydra serpentina (Phillips et al., 1996) acknowledged four subspecies, C. s. serpentina (Northern U.S. and Figure 2. Side profile of Chelydra serpentina. Note Canada), C. s. osceola (Southeastern U.S.), C. s. the serrated posterior end of the carapace and the rossignonii (Central America), and C. s. tail’s raised central ridge. Photo from http://pelotes.jea.com/AnimalFact/Reptile/snapturt.ht acutirostris (South America). Recent IUCN m. reclassification of chelonians based on genetic analyses (Rhodin et al., 2010) elevated C. s. rossignonii and C. s. acutirostris to species level and established C. s. osceola as a synonym for C. s. serpentina, thus eliminating subspecies within C. serpentina. Antiquated distinctions between the two formerly recognized North American subspecies were based on negligible morphometric variations between the two populations. Interbreeding in the overlapping range of the two populations was well documented, further discrediting the validity of the subspecies distinction (Feuer, 1971; Aresco and Gunzburger, 2007). Therefore, any emphasis of subspecies differentiation in the ensuing literature should be disregarded. Figure 3. Front-view of a captured Chelydra Continued usage of invalid subspecies names is serpentina. Different skin textures and the distinctive pink mouth are visible from this angle. Photo from still prevalent in the exotic pet trade for C. -

UC Davis UC Davis Previously Published Works
UC Davis UC Davis Previously Published Works Title Survival, Demography, and Growth of Gopher Tortoises (Gopherus polyphemus) from Three Study Sites with Different Management Histories Permalink https://escholarship.org/uc/item/9sc747h0 Journal JOURNAL OF WILDLIFE MANAGEMENT, 78(7) ISSN 0022-541X Authors Tuberville, Tracey D Todd, Brian D Hermann, Sharon M et al. Publication Date 2014-09-01 DOI 10.1002/jwmg.773 Peer reviewed eScholarship.org Powered by the California Digital Library University of California The Journal of Wildlife Management 78(7):1151–1160; 2014; DOI: 10.1002/jwmg.773 Research Article Survival, Demography, and Growth of Gopher Tortoises (Gopherus polyphemus) from Three Study Sites with Different Management Histories TRACEY D. TUBERVILLE,1 University of Georgia’s Savannah River Ecology Lab, Drawer E, Aiken, SC 29802, USA BRIAN D. TODD, Department of Wildlife, Fish, and Conservation Biology, University of California, Davis, One Shields Ave, Davis, CA 95616, USA SHARON M. HERMANN, Department of Biological Sciences, Auburn University, Auburn, AL 36849, USA WILLIAM K. MICHENER, University Libraries, 1312 Basehart SE, Albuquerque, NM 87106, USA CRAIG GUYER, Department of Biological Sciences, Auburn University, Auburn, AL 36849, USA ABSTRACT Recovery or sustainable management of wildlife populations often entails management of habitat on which they depend. In this regard, turtles pose unique conservation challenges because of their life histories. The combination of late maturity, low survival when young, and dependence on high adult survival suggests they may be slow to respond demographically to conventional habitat management. Thus, long- term studies are necessary to understand population dynamics and recovery potential in these species.