Breakdown of the Reciprocal Stabilization of QBRICK Frem1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Global Analysis Reveals the Complexity of the Human Glomerular Extracellular Matrix
Global analysis reveals the complexity of the human glomerular extracellular matrix Rachel Lennon,1,2 Adam Byron,1,* Jonathan D. Humphries,1 Michael J. Randles,1,2 Alex Carisey,1 Stephanie Murphy,1,2 David Knight,3 Paul E. Brenchley,2 Roy Zent,4,5 and Martin J. Humphries.1 1Wellcome Trust Centre for Cell-Matrix Research, Faculty of Life Sciences, University of Manchester, Manchester, UK; 2Faculty of Medical and Human Sciences, University of Manchester, Manchester, UK; 3Biological Mass Spectrometry Core Facility, Faculty of Life Sciences, University of Manchester, Manchester, UK; 4Division of Nephrology, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN, USA; and 5Veterans Affairs Hospital, Nashville, TN, USA. *Present address: Edinburgh Cancer Research UK Centre, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh, UK. Running title: Proteome of the glomerular matrix Word count: Abstract: 208, main text 2765 Corresponding author: Dr Rachel Lennon, Wellcome Trust Centre for Cell-Matrix Research, Michael Smith Building, University of Manchester, Manchester M13 9PT, UK. Phone: 0044 (0) 161 2755498. Fax: 0044 (0) 161 2755082. Email: [email protected] Abstract The glomerulus contains unique cellular and extracellular matrix (ECM) components, which are required for intact barrier function. Studies of the cellular components have helped to build understanding of glomerular disease; however, the full composition and regulation of glomerular ECM remains poorly understood. Here, we employed mass spectrometry–based proteomics of enriched ECM extracts for a global analysis of human glomerular ECM in vivo and identified a tissue-specific proteome of 144 structural and regulatory ECM proteins. This catalogue includes all previously identified glomerular components, plus many new and abundant components. -

Propranolol-Mediated Attenuation of MMP-9 Excretion in Infants with Hemangiomas
Supplementary Online Content Thaivalappil S, Bauman N, Saieg A, Movius E, Brown KJ, Preciado D. Propranolol-mediated attenuation of MMP-9 excretion in infants with hemangiomas. JAMA Otolaryngol Head Neck Surg. doi:10.1001/jamaoto.2013.4773 eTable. List of All of the Proteins Identified by Proteomics This supplementary material has been provided by the authors to give readers additional information about their work. © 2013 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 10/01/2021 eTable. List of All of the Proteins Identified by Proteomics Protein Name Prop 12 mo/4 Pred 12 mo/4 Δ Prop to Pred mo mo Myeloperoxidase OS=Homo sapiens GN=MPO 26.00 143.00 ‐117.00 Lactotransferrin OS=Homo sapiens GN=LTF 114.00 205.50 ‐91.50 Matrix metalloproteinase‐9 OS=Homo sapiens GN=MMP9 5.00 36.00 ‐31.00 Neutrophil elastase OS=Homo sapiens GN=ELANE 24.00 48.00 ‐24.00 Bleomycin hydrolase OS=Homo sapiens GN=BLMH 3.00 25.00 ‐22.00 CAP7_HUMAN Azurocidin OS=Homo sapiens GN=AZU1 PE=1 SV=3 4.00 26.00 ‐22.00 S10A8_HUMAN Protein S100‐A8 OS=Homo sapiens GN=S100A8 PE=1 14.67 30.50 ‐15.83 SV=1 IL1F9_HUMAN Interleukin‐1 family member 9 OS=Homo sapiens 1.00 15.00 ‐14.00 GN=IL1F9 PE=1 SV=1 MUC5B_HUMAN Mucin‐5B OS=Homo sapiens GN=MUC5B PE=1 SV=3 2.00 14.00 ‐12.00 MUC4_HUMAN Mucin‐4 OS=Homo sapiens GN=MUC4 PE=1 SV=3 1.00 12.00 ‐11.00 HRG_HUMAN Histidine‐rich glycoprotein OS=Homo sapiens GN=HRG 1.00 12.00 ‐11.00 PE=1 SV=1 TKT_HUMAN Transketolase OS=Homo sapiens GN=TKT PE=1 SV=3 17.00 28.00 ‐11.00 CATG_HUMAN Cathepsin G OS=Homo -

Environmental Influences on Endothelial Gene Expression
ENDOTHELIAL CELL GENE EXPRESSION John Matthew Jeff Herbert Supervisors: Prof. Roy Bicknell and Dr. Victoria Heath PhD thesis University of Birmingham August 2012 University of Birmingham Research Archive e-theses repository This unpublished thesis/dissertation is copyright of the author and/or third parties. The intellectual property rights of the author or third parties in respect of this work are as defined by The Copyright Designs and Patents Act 1988 or as modified by any successor legislation. Any use made of information contained in this thesis/dissertation must be in accordance with that legislation and must be properly acknowledged. Further distribution or reproduction in any format is prohibited without the permission of the copyright holder. ABSTRACT Tumour angiogenesis is a vital process in the pathology of tumour development and metastasis. Targeting markers of tumour endothelium provide a means of targeted destruction of a tumours oxygen and nutrient supply via destruction of tumour vasculature, which in turn ultimately leads to beneficial consequences to patients. Although current anti -angiogenic and vascular targeting strategies help patients, more potently in combination with chemo therapy, there is still a need for more tumour endothelial marker discoveries as current treatments have cardiovascular and other side effects. For the first time, the analyses of in-vivo biotinylation of an embryonic system is performed to obtain putative vascular targets. Also for the first time, deep sequencing is applied to freshly isolated tumour and normal endothelial cells from lung, colon and bladder tissues for the identification of pan-vascular-targets. Integration of the proteomic, deep sequencing, public cDNA libraries and microarrays, delivers 5,892 putative vascular targets to the science community. -

Original Article FREM2 Is an Independent Predictor of Poor Survival in Clear Cell Renal Cell Carcinoma-Evidence from the Cancer Genome Atlas (TCGA)
Int J Clin Exp Med 2019;12(12):13741-13748 www.ijcem.com /ISSN:1940-5901/IJCEM0076963 Original Article FREM2 is an independent predictor of poor survival in clear cell renal cell carcinoma-evidence from the cancer genome atlas (TCGA) Weiping Huang, Yongyong Lu, Xixi Huang, Feng Wang, Zhixian Yu Department of Urology, The First Affiliated Hospital of Wenzhou Medical University, Wenzhou 325035, Zhejiang Province, China Received February 26, 2018; Accepted October 7, 2018; Epub December 15, 2019; Published December 30, 2019 Abstract: Fraser syndrome protein 1 (FRAS1) and FRAS1 related extracellular matrix protein 1 and 2 (FREM1, FREM2) are a novel group of basement membrane proteins. The relationship between the three gene (FRAS1, FREM1, FREM2) and renal clear cell carcinoma is completely unclear. Thus, in this research, we used the mRNA sequencing data derived from TCGA kidney renal clear cell carcinoma cohort to assess the association of FRAS1, FREM1 and FREM2 with different clinical features. FRAS1, FREM1 and FREM2 mRNA levels were downregulated in KIRC (kidney renal clear cell carcinoma) tissues than normal tissues (FRAS1, P < 0.0001; FREM1, P < 0.0001, FREM2, P = 0.0001), respectively. FRAS1, FREM1 and FREM2 were significantly different in histologic grade, patho- logic stage and pathologic T (all P < 0.001). Low FRAS1, FREM1 and FREM2 expression were correlated to worsen overall survival (all P < 0.01), and Low FREM1 and FREM2 expression had worse relapse-free survival (FREM1, P = 0.0113; FREM2, P = 0.0424). Multivariate Cox regression analysis revealed that FREM2 was an independent prog- nostic factor for overall survival. Taken together, FREM2 expression is an independent predictor of poor survival in renal clear cell carcinoma and is positively associated with advanced stage, high histologic grade. -

Figure S1. Representative Report Generated by the Ion Torrent System Server for Each of the KCC71 Panel Analysis and Pcafusion Analysis
Figure S1. Representative report generated by the Ion Torrent system server for each of the KCC71 panel analysis and PCaFusion analysis. (A) Details of the run summary report followed by the alignment summary report for the KCC71 panel analysis sequencing. (B) Details of the run summary report for the PCaFusion panel analysis. A Figure S1. Continued. Representative report generated by the Ion Torrent system server for each of the KCC71 panel analysis and PCaFusion analysis. (A) Details of the run summary report followed by the alignment summary report for the KCC71 panel analysis sequencing. (B) Details of the run summary report for the PCaFusion panel analysis. B Figure S2. Comparative analysis of the variant frequency found by the KCC71 panel and calculated from publicly available cBioPortal datasets. For each of the 71 genes in the KCC71 panel, the frequency of variants was calculated as the variant number found in the examined cases. Datasets marked with different colors and sample numbers of prostate cancer are presented in the upper right. *Significantly high in the present study. Figure S3. Seven subnetworks extracted from each of seven public prostate cancer gene networks in TCNG (Table SVI). Blue dots represent genes that include initial seed genes (parent nodes), and parent‑child and child‑grandchild genes in the network. Graphical representation of node‑to‑node associations and subnetwork structures that differed among and were unique to each of the seven subnetworks. TCNG, The Cancer Network Galaxy. Figure S4. REVIGO tree map showing the predicted biological processes of prostate cancer in the Japanese. Each rectangle represents a biological function in terms of a Gene Ontology (GO) term, with the size adjusted to represent the P‑value of the GO term in the underlying GO term database. -

Supplemental Material Placed on This Supplemental Material Which Has Been Supplied by the Author(S) J Med Genet
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) J Med Genet Supplement Supplementary Table S1: GENE MEAN GENE NAME OMIM SYMBOL COVERAGE CAKUT CAKUT ADTKD ADTKD aHUS/TMA aHUS/TMA TUBULOPATHIES TUBULOPATHIES Glomerulopathies Glomerulopathies Polycystic kidneys / Ciliopathies Ciliopathies / kidneys Polycystic METABOLIC DISORDERS AND OTHERS OTHERS AND DISORDERS METABOLIC x x ACE angiotensin-I converting enzyme 106180 139 x ACTN4 actinin-4 604638 119 x ADAMTS13 von Willebrand cleaving protease 604134 154 x ADCY10 adenylate cyclase 10 605205 81 x x AGT angiotensinogen 106150 157 x x AGTR1 angiotensin II receptor, type 1 106165 131 x AGXT alanine-glyoxylate aminotransferase 604285 173 x AHI1 Abelson helper integration site 1 608894 100 x ALG13 asparagine-linked glycosylation 13 300776 232 x x ALG9 alpha-1,2-mannosyltransferase 606941 165 centrosome and basal body associated x ALMS1 606844 132 protein 1 x x APOA1 apolipoprotein A-1 107680 55 x APOE lipoprotein glomerulopathy 107741 77 x APOL1 apolipoprotein L-1 603743 98 x x APRT adenine phosphoribosyltransferase 102600 165 x ARHGAP24 Rho GTPase-Activation protein 24 610586 215 x ARL13B ADP-ribosylation factor-like 13B 608922 195 x x ARL6 ADP-ribosylation factor-like 6 608845 215 ATPase, H+ transporting, lysosomal V0, x ATP6V0A4 605239 90 subunit a4 ATPase, H+ transporting, lysosomal x x ATP6V1B1 192132 163 56/58, V1, subunit B1 x ATXN10 ataxin -

Chemical Agent and Antibodies B-Raf Inhibitor RAF265
Supplemental Materials and Methods: Chemical agent and antibodies B-Raf inhibitor RAF265 [5-(2-(5-(trifluromethyl)-1H-imidazol-2-yl)pyridin-4-yloxy)-N-(4-trifluoromethyl)phenyl-1-methyl-1H-benzp{D, }imidazol-2- amine] was kindly provided by Novartis Pharma AG and dissolved in solvent ethanol:propylene glycol:2.5% tween-80 (percentage 6:23:71) for oral delivery to mice by gavage. Antibodies to phospho-ERK1/2 Thr202/Tyr204(4370), phosphoMEK1/2(2338 and 9121)), phospho-cyclin D1(3300), cyclin D1 (2978), PLK1 (4513) BIM (2933), BAX (2772), BCL2 (2876) were from Cell Signaling Technology. Additional antibodies for phospho-ERK1,2 detection for western blot were from Promega (V803A), and Santa Cruz (E-Y, SC7383). Total ERK antibody for western blot analysis was K-23 from Santa Cruz (SC-94). Ki67 antibody (ab833) was from ABCAM, Mcl1 antibody (559027) was from BD Biosciences, Factor VIII antibody was from Dako (A082), CD31 antibody was from Dianova, (DIA310), and Cot antibody was from Santa Cruz Biotechnology (sc-373677). For the cyclin D1 second antibody staining was with an Alexa Fluor 568 donkey anti-rabbit IgG (Invitrogen, A10042) (1:200 dilution). The pMEK1 fluorescence was developed using the Alexa Fluor 488 chicken anti-rabbit IgG second antibody (1:200 dilution).TUNEL staining kits were from Promega (G2350). Mouse Implant Studies: Biopsy tissues were delivered to research laboratory in ice-cold Dulbecco's Modified Eagle Medium (DMEM) buffer solution. As the tissue mass available from each biopsy was limited, we first passaged the biopsy tissue in Balb/c nu/Foxn1 athymic nude mice (6-8 weeks of age and weighing 22-25g, purchased from Harlan Sprague Dawley, USA) to increase the volume of tumor for further implantation. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -

Nephronectin As a Matrix Effector in Cancer
cancers Review Nephronectin as a Matrix Effector in Cancer Synnøve Norvoll Magnussen 1,* , Jimita Toraskar 2,3 , Elin Hadler-Olsen 1,4, Tonje S. Steigedal 2,3 and Gunbjørg Svineng 1 1 Department of Medical Biology, Faculty of Health Sciences, UiT—The Arctic University of Norway, 9037 Tromsø, Norway; [email protected] (E.H.-O.); [email protected] (G.S.) 2 Department of Clinical and Molecular Medicine, Faculty of Medicine and Health Sciences, Norwegian University of Science and Technology (NTNU), 7491 Trondheim, Norway; [email protected] (J.T.); [email protected] (T.S.S.) 3 Cancer Clinic, St. Olavs Hospital HF, 7006 Trondheim, Norway 4 The Public Dental Health Service Competence Center of Northern Norway, 9271 Tromsø, Norway * Correspondence: [email protected] Simple Summary: The extracellular matrix provides an important scaffold for cells and tissues of multicellular organisms. The scaffold not only provides a secure anchorage point, but also functions as a reservoir for signalling molecules, sequestered and released when necessary. A dysregulated extracellular matrix may therefore modulate cellular behaviour, as seen during cancer progression. The extracellular matrix protein nephronectin was discovered two decades ago and found to regulate important embryonic developmental processes. Loss of either nephronectin or its receptor, integrin α8β1, leads to underdeveloped kidneys. Recent findings show that nephronectin is also dysregulated in breast cancer and plays a role in promoting metastasis. To enable therapeutic intervention, it is important to fully understand the role of nephronectin and its receptors in cancer progression. In Citation: Magnussen, S.N.; this review, we summarise the literature on nephronectin, analyse the structure and domain-related Toraskar, J.; Hadler-Olsen, E.; functions of nephronectin and link these functions to potential roles in cancer progression. -

Supplementary Table S5. List of All the Peptides That Were Included in the Peptide Biomarker Panels for Detecting Primary UBC
Supplementary Table S5. List of all the peptides that were included in the peptide biomarker panels for detecting primary UBC Sequence information Peptide- SwissProt Amino acid Sequence‡ Protein name ID† Accession ID 1747 DCGDNSDEEN Low-density lipoprotein receptor-related protein 2 LRP2 1907 SAYQEAMDIS 14-3-3 protein sigma, SFN SFN 1944 PpGPpGpPGPpS Collagen alpha-1(I) chain CO1A1 2032 GGpGSDGKpGPpG Collagen alpha-1(III) chain CO3A1 2200 TPEEKSAVTAL Hemoglobin subunit beta HBA 2451 PTALGVRGASRS Bromodomain-containing protein 1 BRD1 2522 KPLKEPGLGQL Mitogen-activated protein kinase 14 MAPK14 2543 LSALEEYTKK Apolipoprotein A-I APOA1 2592 DDGEAGKpGRpG Collagen alpha-1(I) chain CO1A1 2597 TCGDSCDQCCPG Laminin subunit alpha-2 LAMA2 2794 DGPAGApGTPGpQG Collagen alpha-1(I) chain COL1A1 Basement membrane-specific heparan sulfate 2801 GTCSGCNCNGHAS HSPG2 proteoglycan core protein 2830 VDEVGGEALGRL Hemoglobin subunit beta HBB 2858 EAGGGSNSLQNSP FERM domain-containing protein 4A FRM4A 3183 KPWAAQDGPKPG Leucine-rich repeat-containing protein 25 LRC25 3202 EEYTKKLNTQ Apolipoprotein A-I APOA1 3223 PGYTGNGYGPNGC Cubilin CUBN 3430 KVVAGVANALAHK Hemoglobin subunit delta HBD 3651 MYVSGpPGpPGpP Collagen alpha-1(XVII) chain, COL17A1 COL17A1 3758 VHLTPEEKSAVT Hemoglobin subunit beta HBB 4152 4157 VAGVANALAHKYH Hemoglobin subunit beta HBB 4270 AVADTRDQADGSR Polymeric immunoglobulin receptor PIGR 4532 GSYNLPSLPDIDC Transcription intermediary factor 1-alpha, TRIM24 TRIM24 4535 GPPGpPGPpGPPGPPS Collagen alpha-1(I) chain CO1A1 4759 GpPGEGLPGPpGpPGS Collagen alpha-1(XVII) -

Supporting Information
Supporting Information O’Connell et al. 10.1073/pnas.1109493108 SI Materials and Methods software. For quantification of microvessel density, regions of Cells and Transgenic Mice. 4T1 mammary carcinoma cells and CT26 highest vessel density were first identified at low magnification colorectal cancer cells were grown in DMEM supplemented with (magnification of 10×). Then, under high magnification (mag- 10% (vol/vol) FBS and 100 U/mL penicillin/streptomycin. The nification of 40×), microvessel density was determined by + S100A4-GFP (1), S100A4-tk (2), Tenascin-C KO (3), S100A4-Cre counting CD31 blood vessels in 10 or more visual fields. All flox/flox (4), VEGF-A (5), and Rosa-lox-Stop-lox-YFP (6) transgenic patient samples were processed according to institutional mice have been described elsewhere. guidelines and an institutional review board-approved protocol. + Immunostaining. Harvested tumors and lungs were fixed in 10% Microarray Analysis. S100A4 stromal cells were isolated by (vol/vol) neutral buffered formalin, dehydrated, and embedded fluorescence-activated cell sorting (FACS) from normal lung in paraffin. Antigen retrieval was performed on deparaffinized tissue and metastatic lung tissue of S100A4-GFP mice. RNA was sections with 10 mM citrate buffer (pH 6.0) before im- isolated using the Qiagen RNeasy Plus Micro Kit following munostaining. Alternatively, harvested tumors and lungs were the manufacturer’s instructions and was then amplified with the embedded in O.C.T. medium (TissueTek) and immunostaining NuGen WT-Ovation Pico Kit. Microarray hybridization to the was performed on frozen sections. Anti-tk antibody was obtained Illumina Mouse Ref-8 Expression BeadChip was performed by from William C. -
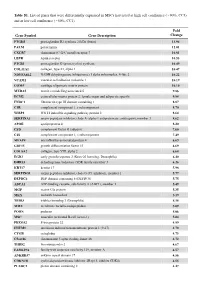
Table S1. List of Genes That Were Differentially Expressed in Mscs Harvested at High Cell Confluence (~90%, CC3) and at Low Cell Confluence (~50%, CC1)
Table S1. List of genes that were differentially expressed in MSCs harvested at high cell confluence (~90%, CC3) and at low cell confluence (~50%, CC1). Fold Gene Symbol Gene Description Change PTGDS prostaglandin D2 synthase 21kDa (brain) 13.94 PALM paralemmin 12.01 CXCR7 chemokine (C-X-C motif) receptor 7 10.81 LEPR leptin receptor 10.53 PTGIS prostaglandin I2 (prostacyclin) synthase 10.49 COL11A1 collagen, type XI, alpha 1 10.47 NDUFA4L2 NADH dehydrogenase (ubiquinone) 1 alpha subcomplex, 4-like 2 10.22 VCAM1 vascular cell adhesion molecule 1 10.19 COMP cartilage oligomeric matrix protein 10.10 MXRA5 matrix-remodelling associated 5 9.86 ECM2 extracellular matrix protein 2, female organ and adipocyte specific 9.84 FNDC1 fibronectin type III domain containing 1 8.87 C1R complement component 1, r subcomponent 8.70 WISP2 WNT1 inducible signaling pathway protein 2 8.64 SERPINA3 serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 3 8.62 APOE apolipoprotein E 8.20 CFD complement factor D (adipsin) 7.60 C1S complement component 1, s subcomponent 7.49 MFAP4 microfibrillar-associated protein 4 6.69 GDF15 growth differentiation factor 15 6.69 COL8A2 collagen, type VIII, alpha 2 6.68 EGR2 early growth response 2 (Krox-20 homolog, Drosophila) 6.40 DHRS3 dehydrogenase/reductase (SDR family) member 3 6.26 KRT17 keratin 17 5.98 SERPING1 serpin peptidase inhibitor, clade G (C1 inhibitor), member 1 5.77 DEPDC6 DEP domain containing 6 (DEPDC6) 5.75 ABCA1 ATP-binding cassette, sub-family A (ABC1), member 1 5.49 MGP matrix