Smoking Dysregulates the Human Airway Basal Cell Transcriptome at COPD Risk Locus 19Q13.2
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mouse Rabac1 Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Rabac1 Knockout Project (CRISPR/Cas9) Objective: To create a Rabac1 knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Rabac1 gene (NCBI Reference Sequence: NM_010261 ; Ensembl: ENSMUSG00000003380 ) is located on Mouse chromosome 7. 5 exons are identified, with the ATG start codon in exon 1 and the TAA stop codon in exon 5 (Transcript: ENSMUST00000076961). Exon 1~5 will be selected as target site. Cas9 and gRNA will be co-injected into fertilized eggs for KO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 1 starts from about 0.18% of the coding region. Exon 1~5 covers 100.0% of the coding region. The size of effective KO region: ~2657 bp. The KO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele 5' gRNA region gRNA region 3' 1 2 3 4 5 Legends Exon of mouse Rabac1 Knockout region Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot (up) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section upstream of start codon is aligned with itself to determine if there are tandem repeats. No significant tandem repeat is found in the dot plot matrix. So this region is suitable for PCR screening or sequencing analysis. Overview of the Dot Plot (down) Window size: 15 bp Forward Reverse Complement Sequence 12 Note: The 2000 bp section downstream of stop codon is aligned with itself to determine if there are tandem repeats. -

Nuclear and Mitochondrial Genome Defects in Autisms
UC Irvine UC Irvine Previously Published Works Title Nuclear and mitochondrial genome defects in autisms. Permalink https://escholarship.org/uc/item/8vq3278q Journal Annals of the New York Academy of Sciences, 1151(1) ISSN 0077-8923 Authors Smith, Moyra Spence, M Anne Flodman, Pamela Publication Date 2009 DOI 10.1111/j.1749-6632.2008.03571.x License https://creativecommons.org/licenses/by/4.0/ 4.0 Peer reviewed eScholarship.org Powered by the California Digital Library University of California THE YEAR IN HUMAN AND MEDICAL GENETICS 2009 Nuclear and Mitochondrial Genome Defects in Autisms Moyra Smith, M. Anne Spence, and Pamela Flodman Department of Pediatrics, University of California, Irvine, California In this review we will evaluate evidence that altered gene dosage and structure im- pacts neurodevelopment and neural connectivity through deleterious effects on synap- tic structure and function, and evidence that the latter are key contributors to the risk for autism. We will review information on alterations of structure of mitochondrial DNA and abnormal mitochondrial function in autism and indications that interactions of the nuclear and mitochondrial genomes may play a role in autism pathogenesis. In a final section we will present data derived using Affymetrixtm SNP 6.0 microar- ray analysis of DNA of a number of subjects and parents recruited to our autism spectrum disorders project. We include data on two sets of monozygotic twins. Col- lectively these data provide additional evidence of nuclear and mitochondrial genome imbalance in autism and evidence of specific candidate genes in autism. We present data on dosage changes in genes that map on the X chromosomes and the Y chro- mosome. -

Genome-Wide Association Identifies Candidate Genes That Influence The
Genome-wide association identifies candidate genes that influence the human electroencephalogram Colin A. Hodgkinsona,1, Mary-Anne Enocha, Vibhuti Srivastavaa, Justine S. Cummins-Omana, Cherisse Ferriera, Polina Iarikovaa, Sriram Sankararamanb, Goli Yaminia, Qiaoping Yuana, Zhifeng Zhoua, Bernard Albaughc, Kenneth V. Whitea, Pei-Hong Shena, and David Goldmana aLaboratory of Neurogenetics, National Institute on Alcohol Abuse and Alcoholism, Rockville, MD 20852; bComputer Science Department, University of California, Berkeley, CA 94720; and cCenter for Human Behavior Studies, Weatherford, OK 73096 Edited* by Raymond L. White, University of California, Emeryville, CA, and approved March 31, 2010 (received for review July 23, 2009) Complex psychiatric disorders are resistant to whole-genome reflects rhythmic electrical activity of the brain. EEG patterns analysis due to genetic and etiological heterogeneity. Variation in dynamically and quantitatively index cortical activation, cognitive resting electroencephalogram (EEG) is associated with common, function, and state of consciousness. EEG traits were among the complex psychiatric diseases including alcoholism, schizophrenia, original intermediate phenotypes in neuropsychiatry, having been and anxiety disorders, although not diagnostic for any of them. EEG first recorded in humans in 1924 by Hans Berger, who documented traits for an individual are stable, variable between individuals, and the α rhythm, seen maximally during states of relaxation with eyes moderately to highly heritable. Such intermediate phenotypes closed, and supplanted by faster β waves during mental activity. appear to be closer to underlying molecular processes than are EEG can be used clinically for the evaluation and differential di- clinical symptoms, and represent an alternative approach for the agnosis of epilepsy and sleep disorders, differentiation of en- identification of genetic variation that underlies complex psychiat- cephalopathy from catatonia, assessment of depth of anesthesia, ric disorders. -

Gprc5b Modulates Inflammatory Response in Glomerular Diseases
BASIC RESEARCH www.jasn.org GPRC5b Modulates Inflammatory Response in Glomerular Diseases via NF-kB Pathway Sonia Zambrano,1 Katja Möller-Hackbarth,1 Xidan Li,1 Patricia Q. Rodriguez,1 Emmanuelle Charrin,1 Angelina Schwarz,1 Jenny Nyström,2 Annika Östman Wernerson,3 Mark Lal,4 and Jaakko Patrakka1 1Karolinska Insitutet/AstraZeneca Integrated Cardio Metabolic Center, Department of Laboratory Medicine, Karolinska Institutet at Karolinska University Hospital Huddinge, Stockholm, Sweden; 2Department of Physiology, Institute of Neuroscience and Physiology, University of Gothenburg, Gothenburg, Sweden; 3Division of Renal Medicine, Department of Clinical Sciences, Intervention and Technology, Karolinska Institutet, Stockholm, Sweden; and 4Division of Bioscience, Department of Cardiovascular, Renal and Metabolic Diseases, Innovative Medicines Biotech Unit, AstraZeneca, Gothenburg, Sweden ABSTRACT Background Inflammatory processes play an important role in the pathogenesis of glomerulopathies. Finding novel ways to suppress glomerular inflammation may offer a new way to stop disease progression. However, the molecular mechanisms that initiate and drive inflammation in the glomerulus are still poorly understood. Methods We performed large-scale gene expression profiling of glomerulus-associated G protein– coupled receptors (GPCRs) to identify new potential therapeutic targets for glomerulopathies. The expression of Gprc5b in disease was analyzed using quantitative PCR and immunofluorescence, and by analyzing published microarray data sets. In vivo studies were carried out in a podocyte-specificGprc5b knockout mouse line. Mechanistic studies were performed in cultured human podocytes. Results We identified an orphan GPCR, Gprc5b, as a novel gene highly enriched in podocytes that was significantly upregulated in common human glomerulopathies, including diabetic nephropathy, IgA ne- phropathy, and lupus nephritis. Similar upregulation of Gprc5b was detected in LPS-induced nephropathy in mice. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

MBNL1 Regulates Essential Alternative RNA Splicing Patterns in MLL-Rearranged Leukemia
ARTICLE https://doi.org/10.1038/s41467-020-15733-8 OPEN MBNL1 regulates essential alternative RNA splicing patterns in MLL-rearranged leukemia Svetlana S. Itskovich1,9, Arun Gurunathan 2,9, Jason Clark 1, Matthew Burwinkel1, Mark Wunderlich3, Mikaela R. Berger4, Aishwarya Kulkarni5,6, Kashish Chetal6, Meenakshi Venkatasubramanian5,6, ✉ Nathan Salomonis 6,7, Ashish R. Kumar 1,7 & Lynn H. Lee 7,8 Despite growing awareness of the biologic features underlying MLL-rearranged leukemia, 1234567890():,; targeted therapies for this leukemia have remained elusive and clinical outcomes remain dismal. MBNL1, a protein involved in alternative splicing, is consistently overexpressed in MLL-rearranged leukemias. We found that MBNL1 loss significantly impairs propagation of murine and human MLL-rearranged leukemia in vitro and in vivo. Through transcriptomic profiling of our experimental systems, we show that in leukemic cells, MBNL1 regulates alternative splicing (predominantly intron exclusion) of several genes including those essential for MLL-rearranged leukemogenesis, such as DOT1L and SETD1A.Wefinally show that selective leukemic cell death is achievable with a small molecule inhibitor of MBNL1. These findings provide the basis for a new therapeutic target in MLL-rearranged leukemia and act as further validation of a burgeoning paradigm in targeted therapy, namely the disruption of cancer-specific splicing programs through the targeting of selectively essential RNA binding proteins. 1 Division of Bone Marrow Transplantation and Immune Deficiency, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 2 Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. 3 Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH 45229, USA. -

Genetics of Familial Non-Medullary Thyroid Carcinoma (FNMTC)
cancers Review Genetics of Familial Non-Medullary Thyroid Carcinoma (FNMTC) Chiara Diquigiovanni * and Elena Bonora Unit of Medical Genetics, Department of Medical and Surgical Sciences, University of Bologna, 40138 Bologna, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-051-208-8418 Simple Summary: Non-medullary thyroid carcinoma (NMTC) originates from thyroid follicular epithelial cells and is considered familial when occurs in two or more first-degree relatives of the patient, in the absence of predisposing environmental factors. Familial NMTC (FNMTC) cases show a high genetic heterogeneity, thus impairing the identification of pivotal molecular changes. In the past years, linkage-based approaches identified several susceptibility loci and variants associated with NMTC risk, however only few genes have been identified. The advent of next-generation sequencing technologies has improved the discovery of new predisposing genes. In this review we report the most significant genes where variants predispose to FNMTC, with the perspective that the integration of these new molecular findings in the clinical data of patients might allow an early detection and tailored therapy of the disease, optimizing patient management. Abstract: Non-medullary thyroid carcinoma (NMTC) is the most frequent endocrine tumor and originates from the follicular epithelial cells of the thyroid. Familial NMTC (FNMTC) has been defined in pedigrees where two or more first-degree relatives of the patient present the disease in absence of other predisposing environmental factors. Compared to sporadic cases, FNMTCs are often multifocal, recurring more frequently and showing an early age at onset with a worse outcome. FNMTC cases Citation: Diquigiovanni, C.; Bonora, E. -

Genetic Influences on Brain Gene Expression in Rats Selected for Tameness and Aggression
Genetic Influences on Brain Gene Expression in Rats Selected for Tameness and Aggression Henrike O. Heyne*,§, Susann Lautenschläger§, Ronald Nelson†, François Besnier†,‡, Maxime Rotival**, Alexander Cagan*, Rimma Kozhemyakina§§, Irina Z. Plyusnina§§, Lyudmila Trut§§, Örjan Carlborg†, Enrico Petretto**, Leonid Kruglyak††,‡‡,***, Svante Pääbo*, Torsten Schöneberg§, Frank W. Albert*,†† * Department of Evolutionary Genetics, Max Planck Institute for Evolutionary Anthropology, Deutscher Platz 6, 04103 Leipzig, Germany § Institute for Biochemistry, University of Leipzig, Medical Faculty, Johannisallee 30, 04103 Leipzig † Swedish University of Agricultural Sciences, Department of Clinical Sciences, Division of Computational Genetics, Mailing address: Box 7078 SE-75007 Uppsala Sweden ‡ Section of Population Genetics and Ecology, Institute of Marine Research, Bergen, Norway, Havforskningsinstituttet, Postboks 1870, Nordnes 5817, Bergen Norway ** MRC Clinical Sciences Centre, Faculty of Medicine, Imperial College London, Du Cane Road, London W12 0NN, UK §§ Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, 630090 Novosibirsk, Russia †† Department of Human Genetics, University of California, Los Angeles, Gonda Center, 695 Charles E. Young Drive South, Los Angeles, CA 90095, USA ‡‡ Department of Biological Chemistry, University of California, Los Angeles, Gonda Center, 695 Charles E. Young Drive South, Los Angeles, CA 90095, USA *** Howard Hughes Medical Institute, University of California, Los Angeles, Gonda Center, 695 Charles E. Young Drive South, Los Angeles, CA 90095, USA deceased: I.Z.P. Irina Z. Plyusnina . 1 ABSTRACT Inter-individual differences in many behaviors are partly due to genetic differences, but the identification of the genes and variants that influence behavior remains challenging. Here, we studied an F2 intercross of two outbred lines of rats selected for tame and aggressive behavior towards humans for more than 64 generations. -

Multivariate Meta-Analysis of Differential Principal Components Underlying Human Primed and Naive-Like Pluripotent States
bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. October 20, 2020 To: bioRxiv Multivariate Meta-Analysis of Differential Principal Components underlying Human Primed and Naive-like Pluripotent States Kory R. Johnson1*, Barbara S. Mallon2, Yang C. Fann1, and Kevin G. Chen2*, 1Intramural IT and Bioinformatics Program, 2NIH Stem Cell Unit, National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, Maryland 20892, USA Keywords: human pluripotent stem cells; naive pluripotency, meta-analysis, principal component analysis, t-SNE, consensus clustering *Correspondence to: Dr. Kory R. Johnson ([email protected]) Dr. Kevin G. Chen ([email protected]) 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.10.20.347666; this version posted October 21, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. This article is a US Government work. It is not subject to copyright under 17 USC 105 and is also made available for use under a CC0 license. ABSTRACT The ground or naive pluripotent state of human pluripotent stem cells (hPSCs), which was initially established in mouse embryonic stem cells (mESCs), is an emerging and tentative concept. To verify this important concept in hPSCs, we performed a multivariate meta-analysis of major hPSC datasets via the combined analytic powers of percentile normalization, principal component analysis (PCA), t-distributed stochastic neighbor embedding (t-SNE), and SC3 consensus clustering. -

Extrinsic Regulators of Mrna Translation in Developing Brain: Story of Wnts
cells Article Extrinsic Regulators of mRNA Translation in Developing Brain: Story of WNTs Yongkyu Park * , Midori Lofton , Diana Li and Mladen-Roko Rasin * Department of Neuroscience and Cell Biology, Robert Wood Johnson Medical School, Rutgers University, Piscataway, NJ 08854, USA; [email protected] (M.L.); [email protected] (D.L.) * Correspondence: [email protected] (Y.P.); [email protected] (M.-R.R.) Abstract: Extrinsic molecules such as morphogens can regulate timed mRNA translation events in developing neurons. In particular, Wingless-type MMTV integration site family, member 3 (Wnt3), was shown to regulate the translation of Foxp2 mRNA encoding a Forkhead transcription factor P2 in the neocortex. However, the Wnt receptor that possibly mediates these translation events remains unknown. Here, we report Frizzled member 7 (Fzd7) as the Wnt3 receptor that lays downstream in Wnt3-regulated mRNA translation. Fzd7 proteins co-localize with Wnt3 ligands in developing neo- cortices. In addition, the Fzd7 proteins overlap in layer-specific neuronal subpopulations expressing different transcription factors, Foxp1 and Foxp2. When Fzd7 was silenced, we found decreased Foxp2 protein expression and increased Foxp1 protein expression, respectively. The Fzd7 silencing also dis- rupted the migration of neocortical glutamatergic neurons. In contrast, Fzd7 overexpression reversed the pattern of migratory defects and Foxp protein expression that we found in the Fzd7 silencing. We further discovered that Fzd7 is required for Wnt3-induced Foxp2 mRNA translation. Surprisingly, we also determined that the Fzd7 suppression of Foxp1 protein expression is not Wnt3 dependent. In conclusion, it is exhibited that the interaction between Wnt3 and Fzd7 regulates neuronal identity and the Fzd7 receptor functions as a downstream factor in ligand Wnt3 signaling for mRNA translation. -

Genome-Wide Sirna Screen for Mediators of NF-Κb Activation
Genome-wide siRNA screen for mediators SEE COMMENTARY of NF-κB activation Benjamin E. Gewurza, Fadi Towficb,c,1, Jessica C. Marb,d,1, Nicholas P. Shinnersa,1, Kaoru Takasakia, Bo Zhaoa, Ellen D. Cahir-McFarlanda, John Quackenbushe, Ramnik J. Xavierb,c, and Elliott Kieffa,2 aDepartment of Medicine and Microbiology and Molecular Genetics, Channing Laboratory, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115; bCenter for Computational and Integrative Biology, Massachusetts General Hospital, Harvard Medical School, Boston, MA 02114; cProgram in Medical and Population Genetics, The Broad Institute of Massachusetts Institute of Technology and Harvard, Cambridge, MA 02142; dDepartment of Biostatistics, Harvard School of Public Health, Boston, MA 02115; and eDepartment of Biostatistics and Computational Biology and Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, MA 02115 Contributed by Elliott Kieff, December 16, 2011 (sent for review October 2, 2011) Although canonical NFκB is frequently critical for cell proliferation, (RIPK1). TRADD engages TNFR-associated factor 2 (TRAF2), survival, or differentiation, NFκB hyperactivation can cause malig- which recruits the ubiquitin (Ub) E2 ligase UBC5 and the E3 nant, inflammatory, or autoimmune disorders. Despite intensive ligases cIAP1 and cIAP2. CIAP1/2 polyubiquitinate RIPK1 and study, mammalian NFκB pathway loss-of-function RNAi analyses TRAF2, which recruit and activate the K63-Ub binding proteins have been limited to specific protein classes. We therefore under- TAB1, TAB2, and TAB3, as well as their associated kinase took a human genome-wide siRNA screen for novel NFκB activa- MAP3K7 (TAK1). TAK1 in turn phosphorylates IKKβ activa- tion pathway components. Using an Epstein Barr virus latent tion loop serines to promote IKK activity (4). -
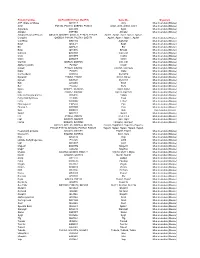
Gene Ids Organism ATP Citrate Synthase Q3V117 Acly
Protein Families UniProtKB ID (from MudPIT) Gene IDs Organism ATP citrate synthase Q3V117 Acly Mus musculus (Mouse) Actin P68134, P60710, Q8BFZ3, P68033 Acta1, Actb, Actbl2, Actc1 Mus musculus (Mouse) Argonaute Q8CJG0 Ago2 Mus musculus (Mouse) Ahnak2 E9PYB0 Ahnak2 Mus musculus (Mouse) Adaptor Related Protein Q8CC13, Q8CBB7, Q3UHJ0, P17426, P17427, Ap1b1, Ap1g1, Aak1, Ap2a1, Ap2a2, Complex Q9DBG3, P84091, P62743, Q9Z1T1 Ap2b1, Ap2m1, Ap2s1 , Ap3b1 Mus musculus (Mouse) V-ATPase Q9Z1G4 Atp6v0a1 Mus musculus (Mouse) Bag3 Q9JLV1 Bag3 Mus musculus (Mouse) Bcr Q6PAJ1 Bcr Mus musculus (Mouse) Bmp Q91Z96 Bmp2k Mus musculus (Mouse) Calcoco Q8CGU1 Calcoco1 Mus musculus (Mouse) Ccdc D3YZP9 Ccdc6 Mus musculus (Mouse) Clint1 Q5SUH7 Clint1 Mus musculus (Mouse) Clathrin Q6IRU5, Q68FD5 Cltb, Cltc Mus musculus (Mouse) Alpha-crystallin P23927 Cryab Mus musculus (Mouse) Casein P19228, Q02862 Csn1s1, Csn1s2a Mus musculus (Mouse) Dab2 P98078 Dab2 Mus musculus (Mouse) Connecdenn Q8K382 Dennd1a Mus musculus (Mouse) Dynamin P39053, P39054 Dnm1, Dnm2 Mus musculus (Mouse) Dynein Q9JHU4 Dync1h1 Mus musculus (Mouse) Edc Q3UJB9 Edc4 Mus musculus (Mouse) Eef P58252 Eef2 Mus musculus (Mouse) Epsin Q80VP1, Q5NCM5 Epn1, Epn2 Mus musculus (Mouse) Eps P42567, Q60902 Eps15, Eps15l1 Mus musculus (Mouse) Fatty acid binding protein Q05816 Fabp5 Mus musculus (Mouse) Fatty Acid Synthase P19096 Fasn Mus musculus (Mouse) Fcho Q3UQN2 Fcho2 Mus musculus (Mouse) Fibrinogen A E9PV24 Fga Mus musculus (Mouse) Filamin A Q8BTM8 Flna Mus musculus (Mouse) Gak Q99KY4 Gak Mus musculus (Mouse)