A Dissertation Entitled Evolutionary Patterns and Occurrences of The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Spice Large.Pdf
Gernot Katzer’s Spice List (http://gernot-katzers-spice-pages.com/engl/) 1/70 (November 2015) Important notice Copyright issues This document is a byproduct of my WWW spice pages. It lists names of spices in about 100 different languages as well as the sci- This document, whether printed or in machine-readable form, may entific names used by botanists and pharmacists, and gives for each be copied and distributed without charge, provided the above no- local name the language where it is taken from and the botanical tice and my address are retained. If the file content (not the layout) name. This index does not tell you whether the plant in question is is modified, this should be indicated in the header. discussed extensively or is just treated as a side-note in the context of another spice article. Employees of Microsoft Corporation are excluded from the Another point to make perfectly clear is that although I give my above paragraph. On all employees of Microsoft Corporation, a best to present only reliable information here, I can take no warrant licence charge of US$ 50 per copy for copying or distributing this of any kind that this file, or the list as printed, or my whole WEB file in all possible forms is levied. Failure to pay this licence charge pages or anything else of my spice collection are correct, harm- is liable to juristical prosecution; please contact me personally for less, acceptable for non-adults or suitable for any specific purpose. details and mode of paying. All other usage restrictions and dis- Remember: Anything free comes without guarantee! claimers decribed here apply unchanged. -
Tourism in Greece 2020 U L B
Travel, Excursions & Sightseeing Tourism in Greece 2020 U L B S O N O K Y M / l e t o c e r G The National Herald T H D E L N A AT ER IONAL H www.thenationalherald.com 2 SPECIAL EDITION JANUARY 2020 Tourism in Greece 2020 THE NATIONAL HERALD ❙ “Greece is a good place for rebirths” Judith Martin Welcome to The National Herald’s first special insert (of many) of 2020! HIS YEAR, we have decided to jumpstart the new urning on summer vacation plans. The Travel Show is To those who have not visited Greece, it is known for Make your decade with an insert focused on various tourist one of the most comprehensive in North America – at - being the birthplace of democracy, the creation of the destinations throughout Greece. We know that as tracting more than 35K attendees and hosting over 750 Olympic Games, and for its unique and historical archi - summer plans winter (finally) sets in here on the east coast of exhibits related to trade and consumer travel. The show tecture. But it is so much more than that. Even the littlest Tthe United States, we Greeks living abroad can’t help it is the ultimate travel and tourism event and aims to pro - moments in Greece have the capacity to fill all your sen - when our minds wander off to our favorite beaches, vil - mote all aspects of tourism worldwide – including local ses at one time. As Henry Miller said, “it takes a lifetime now and lages, and sunsets in the motherland. -
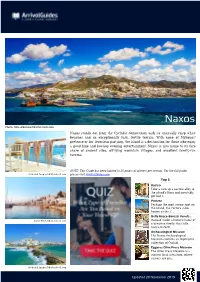
Updated 20 November 2019 Naxos Stands out from the Cycladic Competition with Its Unusually Crisp White Beaches and an Exceptiona
Naxos Photo: Mila Atkovska/Shutterstock.com Naxos stands out from the Cycladic competition with its unusually crisp white beaches and an exceptionally lush, fertile terrain. With none of Mykonos' pretense or Ios' ferocious partying, the island is a destination for those who enjoy a good hike and low-key evening entertainment. Naxos is also home to its fare share of ancient sites, off-lying mountain villages, and excellent family-run taverns. NOTE: This Guide has been limited to 20 points of interest per section. For the full guide, Aetherial Images/Shutterstock.com please visit ArrivalGuides.com Top 5 Kastro Take a turn up a narrow alley in the island's Hora and inevitably get lost i... Portara Perhaps the most scenic spot on the island, the Portara (also known as the T... Della Rocca-Barozzi Veneti... Justin Black/Shutterstock.com Housed inside a historic home of a Venetian family, the Della Rocca-Barozzi ... Archaeological Museum The Naxos Archaeological Museum contains an impressive collection of Cycladi... Eggares Olive Press Museum The Olive Press Museum is a curious local attraction, where visitors are gra... Aetherial Images/Shutterstock.com Updated 20 November 2019 Destination: Naxos Publishing date: 2019-11-20 THE ISLAND BEACHES Mila Atkovska/Shutterstock.com Korpithas/Shutterstock.com Life on Naxos centers around the Hora, or Naxos Naxos is especially well-known for its Town, where most of its taverns, restaurants uncharacteristically beautiful beaches, the most and shopping spots are concentrated. Travelers attractive of which are concentrated along the will need to venture out south of town to nd island's southwestern coast. -

Greece Pre-Departure Guide
GREECE PRE-DEPARTURE GUIDE adventure travel company Early Greek history is the story of internal struggles, from the Mycenaean and Minoan cultures of the Bronze Age to the competing city-states that emerged in the 1st millenium BC. Greece also hosted Alexander the Great’s empire during the Hellenistic period. With the defeat of the Macedonians by the Romans in 168 BC, Greece became the cultural centre of Rome. Greece was also one of the most important cultural and commercial centres during the Byzantine period (4th-15th century AD). Today, Greece is one of the most important travel destinations in the world with a wide collection of historical monuments, ruins, cultural and natural attractions. With more than 2,900 islands and islets, 1,431 beaches, endless blue seas and over 3,700 km of coastline, Greece offers a lot to everyone. Out of 365 days a year, Greece’s sunshine and blue skies are well above 217 days... Each and every of the islands and islets offer unique beauty, charm, great nature, superb beaches, history and adventure. Travel Talk Greece tours take you to the best and most popular Hellas islands including Cyclades (Mykonos, , Ios, Santorini ) islands in great convenience. We hope that our philosophy will exceed your expectations and the following information will help you enjoy a memorable journey in Greece. VISAS It is often difficult to find Theprecise medication EATING & DRINKING that is available in one's home country, so To speak of Greek cuisine will awaken any EU members, as well as citizens of the US, Canada, we recommend that you carry all neccesary Australia, and New Zealand are all automatically medication with you. -

Champagnes and Sparklings White Wines by Style
champagnes and sparklings by bottle by the glass 750ml 150ml Taittinger Champagne Brut, Reserve (chardonnay, pinot noir, pinot meunier) 145.00€ 29.00€ Pierre Gimonnet & Fils, Rose de Blanc, (chardonnay, pinot noir) 120.00€ Karanika Brut, cuvee special, (xinomavro), Amydaio 58.00€ 12.00€ Kir-Yanni Estate, rose, akakies sparkling, (xinomavro), Amydaio 48.00€ 10.00€ Moscato d’asti, pietro forno, il falcetto, piedmonte Italy 38.00€ 9.00€ white wines by style by bottle by the glass Fresh, light bodied and fruity 750ml 150ml Troupis winery, Tomi (moschofilero), P.D.O. Mantineia 36.00€ 7.00€ Alfa Estate, xelones (malagouzia), P.G.I. Florina 39.00€ Plano, Techni Oinou Estate (malagouzia), P.G.I. Drama 38.00€ Karipidis Winery, fume (sauvignon blanc), P.G.I. Kranona 44.00€ Sancere, Jean Paul-Balland (sauvignon blanc), A.O.P. Sancere, France 72.00€ 15.00€ 1827 Navarino Vineyards (chardonnay, roditis), Peloponisos 26.00€ Ktima Dio Ypsi, Dialogos (kydonitsa, asyrtiko) P.G.I. Ilia 38.00€ Pinot Grigio, Santa Margherita, D.O.C. Alto Adige, Italy 42.00€ Gerovasiliou Estate (viognier), P.G.I. Epanomi, Macedonia 64.00€ A-muse, Muses Estate (Gewurstraminer, Malagouzia) P.G.I. Central Gr. 38.00€ Amariotakis Winery, Desmos (Vidiano), P.G.I. Crete 34.00€ 7.00€ Kir-Yianni Estate, Drumo (sauvignon blanc), P.G.I. Florina, Macedonia 62.00€ by bottle by the glass Sharp and crispy, with acidity 750ml 150ml Chablis, Vincent dampt (chardonnay) A.O.P. Chablis, France 72.00€ 14.00€ Artemis Karamolegos (Assyrtiko,Athiri,Aidani) P.D.O. Santorini 62.00€ Two Peaks, Pieter Griebeler (sauvignon blanc), New Zealand 46.00€ Tselepos Winery (Assyrtiko) P.D.O. -

Mindemoya Log - July, 2005
Mindemoya Log - July, 2005 August 1 and we are anchored close to the beach hiding from the meltemi so it is a good day to write the log while the captain tears apart the engine in search of the black-smoke gremlin. The past month has been one of culture. At the beginning of July we sailed for Mykonos and arriving off the south coast enjoyed a lunch and a swim including cleaning of the waterline and propeller before heading for the “EU” marina. Such marinas are plentiful in Greece where the EU monies were used to build the structure but the matching Greek funds never added the utilities or bow mooring lines or management. We walked across the road for a cold drink and watched the filming of a segment of E Television Travelogue that is to air at 10 pm August 8. Maybe you’ll see us at the next table. The next day we were up early to catch a bus to town and a ferry to Delos where we tramped around the ruins for 5 hours learning about the most important archeological site in the Cyclades and the mythical birthplace of the twins Apollo and Artemis. In the 8th century BC a festival in honor of Apollo was established there and the oldest shrine remains are from that era. Delos was important politically and economically thus the many splendid homes of the wealthy. We enjoyed the striking mosaics which are still exquisite art works. The Sanctuary of Apollo contained the temples dedicated to him and the much-photographed Terrace of the Lions. -
Split Board Votes on Teacher Pact
MP •^w. •- Ttfr/i W.t tfeJrtor H rvjl tht fb« c( ywr ObMfttrwrt*;.' OwtouehnWW. (Wttw, *» ft'tri «*}*• W» (o jrt ¢4 r#«pi- pWIWtOfl&W, : VOLUME 28 NUMBF.R 31 MONDAY, SEPTEMBER 28,1992 • WESTLAND, MICHIGAN • 40 PAGES FIFTY CENTS ', 1 Yi2 SutMtao CvT.T*scc»vjt\i CvfK/n<n IN THE PAPER Split board votes on teacher pact Board trustee Fred Warmbier feels that the proposed expected to vote to approve the con not Crosse Pointe." TODAY tract. Board members Fred Warmbier Under the proposed contract, teach teachers' pay raises are too high considering the ecovari d Laurel.Raisanen are expected to ers who have master's degrees and who nomic slump, but most of his colleagues are expected-oppose-it-^—— - .— -, '•••• .--'-- -'•';': .--are at. the top of the_salary_schcduie to ratify a new union contract tonight. ' Early on, teacher union leaders had would earn $52,755 this school year and School taxes: The Livonia Board indicated that Warmbier and Raisanen $55,395 next year. of Education makes no bones BY DARRELL CLEM Newburgh and Wayne^ - had riot only opposed a pay; raise for about its opposition to a "cut and STAFF* WFUTKR The board's decision wilj come after teachers but wanted concessions. Recession cited A sharply divided Wayne-Westland teachers vote today!by secret ballot on Raisanen declined Friday to say how In opposing the raises, Warmbier cap"proposal oh the Nov. 3 school board is expected to cast a split the' tentative contract agreement, she will vote. cited an economic slump, singling put. bqlloti/2A•••••:' \~ . vote tonight (Monday) in approving a reached Sept. -
Southern Aegean an Experience
SPECIAL EDITION APRIL 2019 Southern Aegean An Experience The National Herald www.thenationalherald.com 2 SPECIAL EDITION APRIL 2019 Tribute to the THE NATIONAL HERALD ❙ Southern Aegean Why A Greek Island-Hop is Your Perfect By My Greek Friend Celebration Vacation elebration vacations have become a ri - sing travel trend in recent years, and is - land-hopping packages have proven to be one of the most popular getaways for couples looking to celebrate a milestone anni - versary or birthday in an unforgettable way. CThe Greek islands, with their whitewashed, bo - ugainvillea-covered houses, delicious food, and ho - spitable local culture are arguably some of the best in the world to plan a vacation around. An excellent way to experience them while com - memorating a special event is to book an organized island-hopping package. Here’s why: CONVENIENCE An island-hopping package eliminates the hassle of planning a multi-stop vacation. Instead of spending hours online researching hotels and arranging transportation, travelers simply choose the package that interests them. The company will arrange airport/port/hotel transfers, ferry tickets and hotel reservations; all you’ll need to do is arrive at your meeting place. This leaves you and your lo - ved one more time to celebrate and enjoy your trip and minimizes stress over timetables and other travel details. That being said, organized island-hopping trips offer vacationers flexibility and the chance to ex - planed city squares and beautiful architecture, in - perience the local culture. As opposed to an island cluding an opera house, attest to its history. cruise that keeps passengers on a strict For a couple’s celebration vacation, such variety arrival/departure schedule, island-hopping pac - ensures that every day you and your significant kages allow visitors to wake up on the island, eat other will experience something memorable. -

EGYPT PRE-DEPARTURE GUIDE Adventure Travel Company
EGYPT PRE-DEPARTURE GUIDE adventure travel company Travel Talk tours will take you on a pilgrimage of culture, history, adventure, fun and relaxation. You will visit ancient sites, explore the treasures of Egypt, discover the magnifi ent pyramids and sphinxs, meet local people and partake in numerous activities throughout your Egypt experience. Before you leave for your adventure, it is important that you read this pre-departure information to ensure that your trip runs as smoothly as possible. While we appreciate that you are embarking on an adventure holiday that will produce its own share of surprises, it is also true that forewarned is forearmed. This information is not written with the intention of being anything other than general information that we hope will help you be more prepared for your holiday. The philosophy of Travel Talk is simple: to provide excellent value for money for budget conscious and adventurous travellers without compromising their experiences. We achieve this by providing quality transportation, accommodation, sightseeing and information services. We hope that our philosophy will exceed your expectations and the following information will help you have a memorable journey in Egypt. VISAS Immunisation is generally available in a series of WEATHER three injections over a period of one month (lasts All visitors to Egypt are required to have a visa 3 to 5 years) or a single visit booster dose (lasts Egyptian summers are hot and dry in most of and a passport valid for another six months. Visas only 6 to 12 months). Please consult a medical the country & humid in the Delta and along can be arranged through Egyptian embassies practitioner for advice. -

Athens to Istanbul
Athens to Istanbul Day 1: Arrival Athens (D) Our adventure tour of Greece commences this evening in the cosmopolitan city of Athens with the Parthenon as our backdrop. After our welcome meeting there is the opportunity to sample famous Greek cuisine before perhaps a stroll to the hill of the Areopagus (the Rock of Ares) for a breathtaking view of Athens by night. Day 2: Athens (B) Today we'll travel back in time to Ancient Greece with a walking tour of Athens. Feel the spark of Greek civilization as you stand beneath the Acropolis; explore the Agora or Marketplace, the beating heart of the ancient Athenian commercial empire; stroll through the Plaka where remnants of both an Ottoman and Venetian past are still in evidence and end the day at the Temple of Olympian Zeus, a structure that took three different dynasties and over 600 years to complete. Day 3: Athens – Santorini (B) (L) This morning we head to the port and board our ferry to Santorini, the southern most island of the Cyclades. Be sure to head to the top deck of the ferry as we approach the caldera (volcanic rim of the island) for some spectacular views. Once we've checked into our hotel we'll head into town for an orientation walking tour before heading to one of the islands best wineries for an afternoon of winetasting. Day 4: Santorini (B) (D) What better way to experience all the spectacular sights that this island has to offer than with a quad bike tour around the island? Quad bikes aren't just for the kids adults of all ages will love exploring the island on our guided tour! We visit the ancient site of Akrotiri, a Minoan Bronze Age settlement, before taking in the breath-taking vista of Red Beach, so named for its' striking slabs of red and black volcanic rock. -

Armenian Food Guide for Tourist
ARMENIAN FOOD GUIDE FOR TOURIST YEREVAN 2001 PREPARED BY DR.ARMEN MEHRABYAN ARE YOU HUNGRY? Dukh sovats eq? In Armenia most relations eventually settle on a feast. Armenians eat often and well. They are very hospitable. When you visit an Armenian family, you must have dinner with them and then compliment the hostess for the delicious meal she has prepared. Every region is famous for certain dishes. The range of dishes and ingredients is huge. You can spend weeks without eating the same dish twice. Eating and being invited to dinner in a typical Armenian family are the most wonderful parts of a visit to Armenia. EATING LIKE A NATIVE helps you order many widely available Armenian dishes. There are literally thousands of dishes to try. Don’t miss out. Armenian cuisine is as ancient as the history of Armenia. It is a wonderful combination of different tastes and aromas. Armenian cuisine is almost wholly indigenous, although, as every cuisine, over time it has adopted several food ideas, cooking methods from different cultures. Closely related to Eastern cuisine, various spices, vegetables, fish and fowl and fruits combine to present a unique experience for any visitor. Armenians use different sorts of fresh and dried herbs, as well as a wide variety of spices. The adjustment and mixtures of flavors, dishes, spices and sweets adopt their variety according to the region they belong. Regional Foods. Most Armenian foods are available throughout the country, but there are some specialties of each region to search out during your visit. West Armenian food (Arevmtyan Hayastany utestner) is famous for its meza (appetizers), abour (soups), aghtzan (salads), meat, fish, fowl as well as meatless dishes (vegetarian), pilaf (noodles), hatz (bread) and anousheghen (dessert). -

Flisvos Beach-Cafè-Restaurant
Menu Flisvos Beach-Cafè-Restaurant With our menu, we invite you to share a selection of combinations of local ingredients, celebrating authentic Naxian flavours. At Flisvos, our farm to table philosophy supports our island farmers & producers with the freshest seasonal organic vegetables & herbs from the Mikri Farma, shepherds’ organic cheeses & butter and free range beef & eggs. A culinary journey with unique tastes all naturally sourced. From our marmalades to our sauces to our delicious desserts, everything is passionately handmade in-house. A warm & friendly gastronomic experience right here on the beach! Coffee & Tea Fresh Juices Espresso Freddo Orange juice Cappuccino Freddo Mixed seasonal fruit juice Frappe Homemade lemonade single double Homemade ginger lemonade Americano Homemade pink lemonade Cappuccino Café latte Espresso Milkshakes We also serve almond or soya milk Vanilla Green tea Chocolate Black tea Strawberry Fresh mint tea Banana Fresh ginger tea (hot or cold) Naxos Mountain tea Soft drinks & Water Beer Soft drinks Alfa (0.33lt) Coca Cola, Coca Cola light and zero Amstel (0.5lt) Fanta orange, lemon and blue, Sprite Alfa Weiss bottle (0.5lt) Schweppes Bitter lemon, Ginger ale, Corona (0,33lt) Tonic, Fuze Ice tea lemon or peach Alfa draught small/big Shandy small/big Alcohol free Amstel (0.33lt) Three Cents Beverages Pink grapefruit soda 56 isles Aegean Wit Ginger beer 56 isles Pilsner Aegean Tonic 56 isles beer, produced in the island of Paros the Mineral water small heart of the “56 isles” of the Cyclades. Mineral water