Production Technologies of CRM from Primary
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Contents 2009
INNEHÅLLSFÖRTECKNING/CONTENTS Page Forssberg, Eric, Luleå University of Technology Energy in mineral beneficiation 1 Acarkan N., Kangal O., Bulut G., Önal G. Istanbul Technical University The comparison of gravity separation and flotation of gold and silver bearing ore 3 Ellefmo, Steinar & Ludvigsen, Erik, Norwegian University of Science and Technology Geological Modelling in a Mineral Resources Management Perspective 15 Hansson, Johan, Sundkvist, Jan-Eric, Bolin, Nils Johan, Boliden Mineral AB A study of a two stage removal process 25 Hulthén, Erik & Evertsson, Magnus, Chalmers University of Technology Optimization of crushing stage using on-line speed control on a cone crusher 37 Hooey1, P.L., Spiller2, D.E, Arvidson3, B.R., Marsden4, P., Olsson5, E., 1MEFOS, 2Eric Spiller Consultants LLC, 3Bo Arvidson Consulting LLC, 4, 5Northland Resources Inc. Metallurgical development of Northland Resources' IOCG resources 47 Ikumapayi, Fatai K., Luleå University of Technology, Sundkvist, Jan-Eric & Bolin, Nils-Johan, Boliden Mineral AB Treatment of process water from molybdenum flotation 65 Johansson, Björn, Boliden Mineral AB Limitations in the flotation process 79 Kongas, Matti & Saloheimo, Kari, Outotec Minerals Oy New innovations in on-stream analysis for flotation circuit management and control 89 Kuyumcu, Halit Z. & Rosenkranz, Jan, TU Berlin Investigation of Fluff Separation from Granulated Waste Plastics to be Used in 99 Blast Furnace Operation Mickelsson1, K-O, Östling2 J, Adolfsson3, G., 1LKAB Malmberget, 2Optimation AB Luleå, 3LKAB Kiruna -

Solvent Extraction of Scandium from Pregnant Leach Solution of Nickel Laterite
SOLVENT EXTRACTION OF SCANDIUM FROM PREGNANT LEACH SOLUTION OF NICKEL LATERITE A THESIS SUBMITTED TO THE GRADUATE SCHOOL OF APPLIED AND NATURAL SCIENCES OF MIDDLE EAST TECHNICAL UNIVERSITY BY ECE FERİZOĞLU IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE IN METALLURGICAL AND MATERIALS ENGINEERING SEPTEMBER 2017 Approval of the thesis: SOLVENT EXTRACTION OF SCANDIUM FROM PREGNANT LEACH SOLUTION OF NICKEL LATERITES submitted by Ece FERİZOĞLU partial fulfillment of the requirements for the degree of Master of Science in Metallurgical and Materials Engineering Department, Middle East Technical University by, Prof. Dr. Gülbin Dural Ünver _________________ Dean, Graduate School of Natural and Applied Sciences Prof. Dr. Hakan Gür _________________ Head of Department, Metallurgical and Materials Engineering Prof. Dr. Rıza Gürbüz _________________ Supervisor, Metallurgical and Materials Engineering Dept., METU Prof. Dr. Yavuz A. Topkaya _________________ Co-Supervisor, Metallurgical and Materials Engineering Dept., METU Examining Committee Members: Prof. Dr. İshak Karakaya _________________ Metallurgical and Materials Engineering Dept., METU Metallurgical and Materials Engineering Dept., METU Prof. Dr. Rıza Gürbüz _________________ Prof. Dr. Yavuz A. Topkaya _________________ Metallurgical and Materials Engineering Dept., METU Assist. Prof. Dr. Metehan ERDOĞAN _________________ Metallurgical and Materials Engineering Dept., Yıldırım Beyazıt University Prof. Dr. Ümit Atalay _________________ Mining Engineering Dept., -

Mineral Processing
Mineral Processing Foundations of theory and practice of minerallurgy 1st English edition JAN DRZYMALA, C. Eng., Ph.D., D.Sc. Member of the Polish Mineral Processing Society Wroclaw University of Technology 2007 Translation: J. Drzymala, A. Swatek Reviewer: A. Luszczkiewicz Published as supplied by the author ©Copyright by Jan Drzymala, Wroclaw 2007 Computer typesetting: Danuta Szyszka Cover design: Danuta Szyszka Cover photo: Sebastian Bożek Oficyna Wydawnicza Politechniki Wrocławskiej Wybrzeze Wyspianskiego 27 50-370 Wroclaw Any part of this publication can be used in any form by any means provided that the usage is acknowledged by the citation: Drzymala, J., Mineral Processing, Foundations of theory and practice of minerallurgy, Oficyna Wydawnicza PWr., 2007, www.ig.pwr.wroc.pl/minproc ISBN 978-83-7493-362-9 Contents Introduction ....................................................................................................................9 Part I Introduction to mineral processing .....................................................................13 1. From the Big Bang to mineral processing................................................................14 1.1. The formation of matter ...................................................................................14 1.2. Elementary particles.........................................................................................16 1.3. Molecules .........................................................................................................18 1.4. Solids................................................................................................................19 -
US Schedule for Internet V2
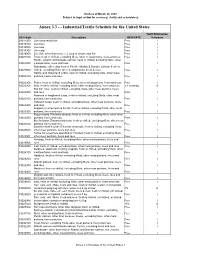
Draft as of March 23, 2007 Subject to legal review for accuracy, clarity and consistency. Annex 3.3 - - Industrial/Textile Schedule for the United States Tariff Elimination US 8 digit Description MFN RATE Schedule 03011000 Live ornamental fish Free I 03019100 Live trout Free I 03019200 Live eels Free I 03019300 Live carp Free I 03019900 Live fish, other than trout, eel, carp or ornamental fish Free I 03021100 Trout, fresh or chilled, excluding fillets, other meat portions, livers and roes Free I Pacific, Atlantic and Danube salmon, fresh or chilled, excluding fillets, other 03021200 meat portions, livers and roes Free I Salmonidae other than trout or Pacific, Atlantic & Danube salmon, fresh or 03021900 chilled, excluding fillets, other meat portions, livers & roes Free I Halibut and Greenland turbot, fresh or chilled, excluding fillets, other meat 03022100 portions, livers and roes Free I 03022200 Plaice, fresh or chilled, excluding fillets, other meat portions, livers and roes Free I 03022300 Sole, fresh or chilled, excluding fillets, other meat portions, livers and roes 1.1 cents/kg A Flat fish, nesi, fresh or chilled, excluding fillets, other meat portions, livers 03022900 and roes Free I Albacore or longfinned tunas, fresh or chilled, excluding fillets, other meat 03023100 portions, livers and roes Free I Yellowfin tunas, fresh or chilled, excluding fillets, other meat portions, livers 03023200 and roes Free I Skipjack or stripe-bellied bonito, fresh or chilled, excluding fillets, other meat 03023300 portions, livers and roes Free -

Media Tracking List Edition January 2021
AN ISENTIA COMPANY Australia Media Tracking List Edition January 2021 The coverage listed in this document is correct at the time of printing. Slice Media reserves the right to change coverage monitored at any time without notification. National National AFR Weekend Australian Financial Review The Australian The Saturday Paper Weekend Australian SLICE MEDIA Media Tracking List January PAGE 2/89 2021 Capital City Daily ACT Canberra Times Sunday Canberra Times NSW Daily Telegraph Sun-Herald(Sydney) Sunday Telegraph (Sydney) Sydney Morning Herald NT Northern Territory News Sunday Territorian (Darwin) QLD Courier Mail Sunday Mail (Brisbane) SA Advertiser (Adelaide) Sunday Mail (Adel) 1st ed. TAS Mercury (Hobart) Sunday Tasmanian VIC Age Herald Sun (Melbourne) Sunday Age Sunday Herald Sun (Melbourne) The Saturday Age WA Sunday Times (Perth) The Weekend West West Australian SLICE MEDIA Media Tracking List January PAGE 3/89 2021 Suburban National Messenger ACT Canberra City News Northside Chronicle (Canberra) NSW Auburn Review Pictorial Bankstown - Canterbury Torch Blacktown Advocate Camden Advertiser Campbelltown-Macarthur Advertiser Canterbury-Bankstown Express CENTRAL Central Coast Express - Gosford City Hub District Reporter Camden Eastern Suburbs Spectator Emu & Leonay Gazette Fairfield Advance Fairfield City Champion Galston & District Community News Glenmore Gazette Hills District Independent Hills Shire Times Hills to Hawkesbury Hornsby Advocate Inner West Courier Inner West Independent Inner West Times Jordan Springs Gazette Liverpool -

Minerals and Metallurgical Engineering
Course Structure B. Tech. - Minerals and Metallurgical Engineering Applicable to those admitted through JEE from 2019 onwards Department of Fuel, Minerals and Metallurgical Engineering Indian Institute of Technology (ISM) Dhanbad Dhanbad, Jharkhand, India (September - 2019) Page 1 of 41 Course Structure SEMESTER III S. Subject Subject Name Lecture Tutorial Practical Credit Contact No. ID (L) (T) (P) Hours Hours 1 FMC201 Colloids and interfacial 3 0 0 9 3 phenomena 2 FMC202 Heat and mass transfer 3 0 0 9 3 3 FMC203 Physical separation 3 0 0 9 3 processes for coal and minerals 4 FME221 Particle technology (ESO 3 0 0 9 3 1) 5 E/SO 2 E/SO 3 0 0 9 3 6 FMC251 Particle technology 0 0 2 2 2 laboratory 7 FMC252 Physical separation 0 0 2 2 2 processes laboratory Total Credit 49 19 SEMESTER IV S. Subject Subject Name Lecture Tutorial Practical Credit Contact No. ID (L) (T) (P) Hours Hours 1 FMC204 Electrochemistry and 3 0 0 9 3 corrosion 2 FMC205 Thermodynamics and 3 0 0 9 3 kinetics 3 FMC206 Phase transformation and 3 0 0 9 3 heat treatment 4 FMC207 Fine particle processing for 3 0 0 9 3 coal and minerals 5 FME222 Introduction to fuel 3 0 0 9 3 technology (ESO 3) 6 FMC253 Fine particle processing 0 0 2 2 2 laboratory 7 FMC254 Introduction to fuel 0 0 2 2 2 technology laboratory Total Credit 49 19 Page 2 of 41 SEMESTER V S. Subject Subject Name Lecture Tutorial Practical Credit Contact No. -

Study of Phosphorus Behaviour in Levitated Silicon-Iron Droplets
STUDY OF PHOSPHORUS BEHAVIOUR IN LEVITATED SILICON-IRON DROPLETS by Katherine Le A thesis submitted in conformity with the requirements for the degree of Master of Applied Science Department of Materials Science and Engineering University of Toronto © Copyright by Katherine Le 2016 ii Study of Phosphorus Behaviour in Levitated Si-Fe Droplets Katherine Le Master of Applied Science Department of Materials Science and Engineering University of Toronto 2016 Abstract While the treatment of relatively inexpensive ferrosilicon alloys is a potential refining route in order to generate solar grade silicon, phosphorus is one of the more difficult impurities to remove by conventional processing. In this project, electromagnetic levitation was used to investigate the dephosphorization of ferrosilicon alloy droplets exposed to H2-Ar gas mixtures under various experimental conditions including, refining time, temperature (1450°C-1720°C), H2-Ar gas concentrations and flow rate, iron alloying content, and initial phosphorus concentration. Reaction rates increased with higher refining times, temperatures, and H2 gas concentrations. With unknown parameters associated with the kinetics of gas phase reactions, the approach involved comparison of apparent activation energies derived for the chemical reaction and gas diffusion steps of the dephosphorization process. The phosphorus removal rate is thought to be controlled by the interfacial reaction step; further work is required to confirm this conclusion. iii Acknowledgements I would like to express my gratitude and respect to my supervisor, Prof. Alex McLean for the opportunity to work on this research. I am thankful for his guidance, wisdom and encouragement throughout the course of my studies. He is a truly inspiring person, and a great enabler of new learning opportunities. -

The Gersdorffite-Bismuthinite-Native Gold Association and the Skarn
minerals Article The Gersdorffite-Bismuthinite-Native Gold Association and the Skarn-Porphyry Mineralization in the Kamariza Mining District, Lavrion, Greece † Panagiotis Voudouris 1,* , Constantinos Mavrogonatos 1 , Branko Rieck 2, Uwe Kolitsch 2,3, Paul G. Spry 4 , Christophe Scheffer 5, Alexandre Tarantola 6 , Olivier Vanderhaeghe 7, Emmanouil Galanos 1, Vasilios Melfos 8 , Stefanos Zaimis 9, Konstantinos Soukis 1 and Adonis Photiades 10 1 Department of Geology & Geoenvironment, National and Kapodistrian University of Athens, 15784 Athens, Greece; [email protected] (C.M.); [email protected] (E.G.); [email protected] (K.S.) 2 Institut für Mineralogie und Kristallographie, Universität Wien, 1090 Wien, Austria; [email protected] 3 Mineralogisch-Petrographische Abteilung, Naturhistorisches Museum, 1010 Wien, Austria; [email protected] 4 Department of Geological and Atmospheric Sciences, Iowa State University, Ames, IA 50011, USA; [email protected] 5 Département de Géologie et de Génie Géologique, Université Laval, Québec, QC G1V 0A6, Canada; [email protected] 6 Université de Lorraine, CNRS, GeoRessources UMR 7359, Faculté des Sciences et Technologies, F-54506 Vandoeuvre-lès-Nancy, France; [email protected] 7 Université de Toulouse, Géosciences Environnement Toulouse (GET), UMR 5563 CNRS, F-31400 Toulouse, France; [email protected] 8 Department of Mineralogy-Petrology-Economic Geology, Faculty of Geology, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece; [email protected] 9 Institut für Mineralogie, TU Bergakademie Freiberg, 09599 Freiberg, Germany; [email protected] 10 Institute of Geology and Mineral Exploration (I.G.M.E.), 13677 Acharnae, Greece; [email protected] * Correspondence: [email protected]; Tel.: +30-210-7274129 † The paper is an extended version of our paper published in 1st International Electronic Conference on Mineral Science. -

Binary and Ternary Transition-Metal Phosphides As Hydrodenitrogenation Catalysts
Research Collection Doctoral Thesis Binary and ternary transition-metal phosphides as hydrodenitrogenation catalysts Author(s): Stinner, Christoph Publication Date: 2001 Permanent Link: https://doi.org/10.3929/ethz-a-004378279 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use. ETH Library Diss. ETH No. 14422 Binary and Ternary Transition-Metal Phosphides as Hydrodenitrogenation Catalysts A dissertation submitted to the Swiss Federal Institute of Technology Zurich for the degree of Doctor of Natural Sciences Presented by Christoph Stinner Dipl.-Chem. University of Bonn born February 27, 1969 in Troisdorf (NRW), Germany Accepted on the recommendation of Prof. Dr. Roel Prins, examiner Prof. Dr. Reinhard Nesper, co-examiner Dr. Thomas Weber, co-examiner Zurich 2001 I Contents Zusammenfassung V Abstract IX 1 Introduction 1 1.1 Motivation 1 1.2 Phosphides 4 1.2.1 General 4 1.2.2 Classification 4 1.2.3 Preparation 5 1.2.4 Properties 12 1.2.5 Applications and Uses 13 1.3 Scope of the Thesis 14 1.4 References 16 2 Characterization Methods 1 2.1 FT Raman Spectroscopy 21 2.2 Thermogravimetric Analysis 24 2.3 Temperature-Programmed Reduction 25 2.4 X-Ray Powder Diffractometry 26 2.5 Nitrogen Adsorption 28 2.6 Solid State Nuclear Magnetic Resonance Spectroscopy 28 2.7 Catalytic Test 33 2.8 References 36 3 Formation, Structure, and HDN Activity of Unsupported Molybdenum Phosphide 37 3.1 Introduction -

Telfer Processing Plant Upgrade – the Implementation of Additional
Telfer Processing Plant Upgrade – The Implementation of Additional Cleaning Capacity and the Regrinding of Copper and Pyrite Concentrates D R Seaman1, F Burns2, B Adamson3, B A Seaman4 and P Manton5 ABSTRACT The Telfer concentrator, located in the Great Sandy Desert of Western Australia, consists of a dual train gold/copper operation processing ore from one underground and, currently, two open pit mines with differing mineralogy. The fl otation circuit of each train was designed to operate in several modes depending on the feed mineralogy. The majority of ore mined at Telfer is processed in a sequential mode where copper minerals are fi rst fl oated into a saleable copper concentrate followed by the fl otation of an auriferous pyrite concentrate which is treated in an on-site hydrometallurgical plant (carbon-in-leach (CIL)). Gold is recovered as a gravity product within the primary grinding circuit, to the copper concentrate, and to a lesser extent, the CIL circuit. Since Telfer was re-opened, with a new concentrator, in 2004, the processing plant has struggled with poor copper concentrate grades, partially due to the excessive entrainment of non-sulfi de gangue minerals in the copper fl otation circuit and, more recently, due to composite copper particles produced when processing ore from a supplementary satellite pit that has not previously been processed through the new Telfer concentrator. Gold recoveries in the CIL circuit have also been below industry standard. This paper presents the implementation of recent changes made to the circuit to address these performance issues. The reconfi guration of the circuit has involved the installation of the following major equipment items: two ISAMills™ in ultra-fi ne grinding applications (one in the copper circuit and one in the pyrite circuit), two Jameson Cells to improve fi ne gangue rejection and a bank of 5 × Outotec TC30s to recovery copper and gold from the reground pyrite stream. -

Xstrata Technology Update Edition 13 – April 2012 Building Plants That Work
xstrata technology update Edition 13 – April 2012 Building plants that work You have to get a lot of things it takes another operator to get them right to build a plant that works. right. Someone who has lived through the problems, had to do the maintenance, operated during a midnight power Of course the big picture must be right – doing the right project, in the right place, failure, cleaned up the spill. Someone at the right time. who has “closed the loop” on previous designs; lived with previous decisions After that, the devil is in the detail. You and improved them, over and over. need a sound design, good execution, good commissioning, and ongoing This is why Xstrata Technology provides support after commissioning. You need a technology “package”. Just as a car to operate and maintain your plant in is more than an engine, technology is the long run, long after the construction more than a single piece of equipment. company has left. That’s when all the Technology is a system. All the elements “little” details become important – how of the system have to work with each easy is it to operate, how good is the other and with the people in the plant. maintenance access, what happens in We want our cars designed by people a power failure, where are the spillage who love cars and driving. So should points and how do we clean them our plants be designed by people with up? Are the instruments reliable and experience and passion to make each is the process control strategy robust one work better than the last. -

Red-Light Leds for Next-Generation Displays 6 July 2020
Red-light LEDs for next-generation displays 6 July 2020 indium gallium nitride (InGaN) semiconductors. Integrating two material systems is difficult. "Creating RGB displays requires the mass transfer of the separate blue, green and red LEDs together," says KAUST researcher Zhe Zhuang. An easier solution would be to create different-colored LEDs all on a single semiconductor chip. Optimizing the geometry, fabrication and electrical contacts is vital to maximizing the efficiency of the LED. Credit: Zhuag et al. Novel red LEDs are more temperature stable than those made using the conventional semiconductor of choice. In efforts to optimize the performance of light- emitting diodes (LEDs), King Abdullah University of Science and Technology researchers are looking at every aspect of the design, fabrication and operation of these devices. Now, they have succeeded in fabricating red LEDs, based on the The team developed an InGaN red LED structure where naturally blue-emitting semiconductor indium the output power is more stable than that of InGaP red gallium nitride, that are as stable as those based LEDs. Credit: Zhe Zhuang on indium gallium phosphide. LEDs are optical sources made from semiconductors that offer improvements on Since InGaP semiconductors are unable to emit conventional visible-light sources in terms of blue or green light, the only solution to making energy saving, smaller size and longer lifetimes. monolithic RGB micro-LEDs is to use InGaN. This LEDs can emit across the spectrum, from the material has the potential to shift its emission from ultraviolet to blue (B), green (G), red (R) and into blue to green, yellow and red by introducing more the infrared.