Development of New Synthetic Methods, Mechanistic Elucidation Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Studies Directed Towards the Stereoselective Total Synthesis of Miyakolide
Studies Directed Towards the Stereoselective Total Synthesis of Miyakolide by Jinhua Song Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Organic Chemistry at the Massachusetts Institute of Technology February, 1999 @1999 Jinhua Song All rights Reserved The author hereby grants MIT permissions to reproduce and to distribute publicly paper and electronic copies of this thesis document in whole or in part. Signature of Author: Department of Chemistry September 25, 1998 Certified by: Professor Satoru Masamune A. C. Cope Professor of Chemistry Thesis Supervisor Accepted by:, ProfessotDietmar Seyferth, Chairman Departmental Committee on Graduate Students MASSACHUSETTS INSTITUTE OF TECHNOLOGY LrL J This doctoral thesis has been examined by a committee of the Department of Chemistry as follows: Professor Timothy M. Swager Chairman Professor Satoru Masamune Thesis Supervisor Professor Rick L. Danheiser , 2 Studies Directed Towards the Stereoselective Total Synthesis of Miyakolide by Jinhua Song Submitted to the Department of Chemistry on September 25, 1998, in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy in Organic Chemistry Abstract Presented are the stereoselective syntheses of the A (C18-C28), B (C14-C17), C (C6-C13), D (Cl-C5), C'D' (C1-C13) fragments and the efficient coupling of B and C'D' fragments of the marine natural product miyakolide, a 24-membered polyketide macrolide which exhibits anti-cancer activity. Fragment A was synthesized from the chiral aldehyde 4-4 through the successful application of the newly developed boron mediated anti-selective aldol methodology using the chiral ester 3-4. -

The Synthesis and Applications of N-Alkenyl Aziridines
The Synthesis and Applications of N-Alkenyl Aziridines by Nicholas A. Afagh A thesis submitted in conformity with the requirements for the degree of Master of Science Department of Chemistry University of Toronto © Copyright by Nicholas A. Afagh 2010 The Synthesis and Applications of N-Alkenyl Aziridines Nicholas A. Afagh Master of Science Department of Chemistry University of Toronto 2010 Abstract N-alkenyl aziridines are a unique class of molecules that do not behave as typical enamines as a result of the inability of the nitrogen atom lone-pair of electrons to delocalize. The attenuated nucleophilicity of these enamines presents opportunities for the selective functionalization and reactivity not available to classical enamines. An operationally simple and mild copper-mediated coupling has been developed that facilitates the preparation of a broad range of N-alkenyl aziridines not available through existing methods. The preparation and reactivity of highly- functionalized N-alkenyl aziridines are reported. Also reported is the application of the chemoselective amine/aldehyde/alkyne (A 3) multicomponent coupling involving amphoteric aziridine aldehydes as the aldehyde component. This coupling allows access to propargyl amines with pendent aziridine functionality. ii Acknowledgments First and foremost, I would like to thank my supervisor, Professor Andrei K. Yudin for his continuous support and encouragement over the past two years. His wealth of knowledge and profound insight into all matters chemistry made for many interesting discussions. In addition, I would like to thank all the members of the Yudin group past and present with whom I have had the distinct pleasure of working alongside and shared many late evenings. -

Effects in Reversible Exothermic Reactions
Effects in reversible exothermic reactions. Effect of Temperature on Equilibrium A temperature change occurs when temperature is increased or decreased by the flow of heat. This shifts chemical equilibria toward the products or reactants, which can be determined by studying the reaction and deciding whether it is endothermic or exothermic. Introduction Le Châtelier's principle states that a change in temperature, pressure, or concentration of reactants in an equilibrated system will stimulate a response that partially off-sets the change to establish a new equilibrium. In the case of changing temperature, adding or removing of heat shifts the equilibrium. However, reactions invariably involve changes in enthalpy, with energy (typically in the form of heat, but can involve light) either being absorbed or released during the reaction. Some chemical reactions -- like burning wood or exploding TNT -- release heat to their surroundings. Chemists call these exothermic reactions. Increasing the temperature affects an exothermic reaction in two different ways: by changing the rate of the reaction and by changing the balance between products and reactants at the end of the reaction. Generally speaking, your reaction will speed up because a higher temperature means more heat and energy in your system. However, in some cases, raising the temperature might shift equilibrium and prevent some of your reaction from occurring Reaction Rates Nearly all reactions go faster as the temperature increases -- exothermic reactions included. The reaction between oxygen in the air and the chemicals in the tip of a match, for example, is so slow at room temperature that nothing seems to happen. When you heat up the tip of the match by striking it against the striker strip on the box, however, the temperature increases and with it the rate of the reaction until it burns with a hot flame. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Development of Novel Peptide Nucleic Acids A
b-Dicarbonyl compounds Compounds having two carbonyl groups separated by an intervening carbon atom are called b-dicarbonyl compounds, and these compounds are highly versatile reagents for organic synthesis. O O O O C C C R C C C OR' b b b-Dicarbonyl system b-Keto ester ** The pKa for such a proton is in the range 9-11, acidic enough to be removed easily by an alkoxide base to form an enolate. O O O H O OR C C C C C.. C + HOR H enolate anion pKa = 9-11 .. .. .. .. .. .. O . O . O O . O O C C C .. C C C R C OR' R C OR' R C OR' H H H Resonance structure of the anion of a b-keto ester Synthesis of b-keto ester (Claisen condensation) O O O NaOC H 2 2 5 CH3COC2H5 CH3CC..HCOC2H5 + C2H5OH + Na (removed by distillation) Sodiumacetoacetic ester HCl O O CH3CCH2COC2H5 Ethyl acetoacetate (acetoacetic ester) (76%) O O O O (1) NaOC2H5 R CH C + H CHC R CH2C CHCOC H + C H OH 2 OC2H5 OC2H5 + 2 5 2 5 (2) H3O R R (R may also be H) b-Keto ester Mechanism Step1 .. O O . .. + . OHC H + C H OH R CHC OC2H5 .. 2 5 RC.. H C OC2H5 2 5 H .. O . RCH C OC2H5 Step 2 .. .. .. .. O . O O O . RCH2C + . RCH2C CH C OC2H5 HC C OC2H5 . C H O . OC2H5 R 2 5 .. R .. O . O .. + . OC2H5 RCH2C CH C OC2H5 .. R Step 3 .. .. O . O H O O . -

Lonza's Chemical Network Diketene and HCN Derivatives, Heterocycles
Lonza’s chemical network Diketene and HCN derivatives, heterocycles and basic chemicals catalog Pharma&Biotech Nutrition Agriculture MaterialsScience PersonalCare Diketene and HCN derivatives, heterocycles and basic chemicals catalog Content About Lonza 3 Introduction 4 Diketene / ketene derivatives 6 Esters 6 Arylides 7 Alkylamides 8 Pyrazolones 9 Dehydroacetic acid 9 Lonzamon monomers 10 Other diketene derivatives 10 HCN derivatives 12 Heterocycles 14 Basic chemicals 16 Others 17 Alphabetical index 18 About Lonza Lonza is the global leader in the production and support of active phar- From 1897 to the present day combining Swiss tradition with global maceutical ingredients both chemically and biotechnologically. Biophar- experience, the company has had an enterprising character, adapting maceuticals are one of the key growth drivers of the pharmaceutical and its offerings and services to the needs of customers and to changing biotechnology industries. technologies. Throughout our history, we have maintained a strong culture of performance, results and dependability that is valued by all Lonza has strong capabilities in large and small molecules, peptides, of our customers. amino acids and niche bioproducts which play an important role in the development of novel medicines and healthcare products. In addition, Lonza’s cracker in Visp is the back bone of a comprehensive fully back- Lonza is a leader in cell-based research, endotoxin detection and cell ward integrated chemical network. Our product portfolio consists of therapy manufacturing. Furthermore, the company is a leading provider HCN- and diketene derivatives as well as basic chemicals which are key of chemical and biotech ingredients to the nutrition, hygiene, preserva- raw materials and intermediates for many sophisticated applications. -

Acetoacetic Ester Synthesis
Programme: B.Sc. B.ed. (Integrated) Course: ORGANIC CHEMISTRY- III Semester: VI Code: CHE-352 Topic: ETHYLACETOACETATEE Date- 07/04/2020 y Only PPe Dr. Angad Kumar Singh ForF Department of Chemistry, Central University of South Bihar, Gaya (Bihar) Note: These materials are only for classroom teaching purpose at Central University of South Bihar. All the data taken from several books, research articles including Wikipedia. Note: These materials are only for classroom teaching purpose at Central University of South Bihar. All the data taken from several books, research articles including Wikipedia. Ethylacetoacetate The Claisen Condensation between esters containing - hdhydrogens, promotdted byabase such as sodium ethoxy ide, toproduce a !- ketoester. One equiv serves as the nucleophilele (enolate)(eno and the other is OnlyO the electrophile which undergoes additiontion andae elimination. The use of stronger bases, e.g. sodium amide or sodiumsodUse hydride instead of sodium ethoxide, often increases the yield.d. al onal Ethylacetoacetate Note: These materials are only for classroom teaching purpose at Central University of South Bihar. All the data taken from several books, research articles including Wikipedia. Mechanism of the Claisen Condensation The reaction is driven to product by the final deprotonation step. Note: These materials are only for classroom teaching purpose at Central University of South Bihar. All the data taken from several books, research articles including Wikipedia. Mixed Claisen Condensation Like mixed aldol reactions, mixed Claisen condensations are useful if differences in reactivity exist betweenn they two esters as for example when one of the esters has no -hydrogenydrogOnlyO . Examples of such esters are: e Use ers excess Note: These materials are only for classroom teaching purpose at Central University of South Bihar. -
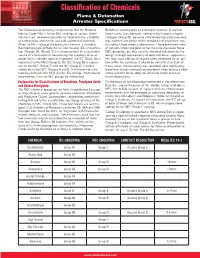
Classification of Chemicals
Classification of Chemicals Flame & Detonation Arrester Specifications PROTECTOSEAL ® The Protectoseal Company recommends that the National Butadiene would qualify as a Group D material. In each of Electric Code (NEC) Article 500, rankings of various chemi - these cases, the chemicals were primarly listed in a higher cals be used, whenever possible, to determine the suitability category (Group B), because of relatively high pressure read - of a detonation arrester for use with a particular chemical. ings noted in one phase of the standard test procedure con - When no NEC rating of the particular chemical is available, ducted by Underwriters Laboratories. These pressures were the International Electrotechnical Commission (IEC) classifica - of concern when categorizing the chemicals because these tion (Groups IIA, IIB and IIC) is recommended as a secondary NEC groupings are also used as standard indicators for the source of information for determining the suitability of an ar - design strength requirements of electrical boxes, apparatus, rester for its intended service. In general, the IEC Group IIA is etc. that must withstand the pressures generated by an igni - equivalent to the NEC Group D; the IEC Group IIB is equiva - tion within the container. It should be noted that, in each of lent to the NEC Group C; and the IEC Group IIC includes these cases, the test pressures recorded were significantly chemicals in the NEC Groups A and B. In the event of a dis - lower than those commonly encountered when testing a deto - crepancy between the NEC and the IEC ratings, Protectoseal nation arrester for its ability to withstand stable and over - recommends that the NEC groups be referenced. -

Ethyl Acetoacetate (Acetoacetic Ester) (B) Diethyl Molonote (Molonic Ester) the Structure of (A) and (B) Are As
PRESENTATION OF ENOLATES Dr. Susmita Bajpai Department of Chemistry B.N.D. College, Kanpur ENOLATES The class of compounds which contain a methylene group (–CH2–) directly bonded to the electron withdrawing groups such as –COCH3, –COOC2H5, –CN, are called active methylene compounds. This is so because the –CH2 group in them is acidic and reactive. The two examples are (a) Ethyl acetoacetate (Acetoacetic ester) (b) Diethyl molonote (Molonic ester) The structure of (a) and (b) are as This reaction is known as claisen condensation Ethyl acetoacetate (CH3COOCH2COOC2H5) • It's IUPAC name is ethyl 3-oxobutanoate • Preparation: Ethyl acetoacetate is prepared by heating ethyl acetate with sodium ethoxide in ethanol, followed by acidification. Claisen condensation • It is a condensation reaction in which, two ester molecules condensed to form an alcohol and a b-Keto ester. Therefore ethyl acetoacetate is a b-Keto ester. • Mechanism: The mechanism involves three steps: + • Step-I :- First sodium ethoxide (C2H5O–Na ) breaks into ethoxide ion and sodium ion. This ethoxide ion attacks ethyl acetoacetate to give ethyl alcohol and ester anion. Step -II: Ester anion attacks the carbonyl group of a second molecules of ethyl acetate. • Step-III: Ethoxide ion is eliminated Properties • It is a colourless pleasant smelling liquid • b.p. - 180.4oC • It is sparingly soluble in water but freely so in organic solvent. • It is neutral to litmus. Chemical properties • It is a tautomeric mixture of Keto and enol forms. Therefore it gives the reaction of the various functional groups present in the two forms. Acidity of methylene hydrogen (Formation of salt) • In ethyl acetoacetate methylene group (–CH2–) flank by two carbonyl group. -

Properties of Silicon Hafensteiner
Baran Lab Properties of Silicon Hafensteiner Si vs. C Siliconium Ion - Si is less electronegative than C - Not believed to exist in any reaction in solution - More facile nucleophilic addition at Si center J. Y. Corey, J. Am. Chem. Soc. 1975, 97, 3237 - Pentacoordinate Si compounds have been observed Average BDE (kcal/mol) MeSiF4 NEt4 Ph3SiF2 NR4 C–C C–Si Si–Si C–F Si–F 83 76 53 116 135 - Lack of cation justified by high rate of bimolecular reactivity at Si C–O Si–O C–H Si–H Mechanism of TMS Deprotection 86 108 83 76 OTMS O Average Bond Lengths (Å) C–C C–Si C–O Si–O 1.54 1.87 1.43 1.66 Workup Si Si Silicon forms weak p-Bonds O O F F NBu4 p - C–C = 65 kcal/mol p - C–Si = 36 kcal/mol Pentavalent Silicon Baran Lab Properties of Silicon Hafensteiner Nucleophilic addition to Si b-Silicon effect and Solvolysis F RO–SiMe3 RO F–SiMe3 SiMe3 Me H H vs. Me3C H Me3C H OSiMe O Li 3 H OCOCF H OCOCF MeLi 3 3 A B Me-SiMe3 12 kA / kB = 2.4 x 10 Duhamel et al. J. Org. Chem. 1996, 61, 2232 H H SiMe Me b-Silicon Effect 3 vs. Me3C H Me3C H - Silicon stabalizes b-carbocations H OCOCF3 H OCOCF3 - Stabalization is a result of hyperconjugation 4 kA / kB = 4 x 10 SiR3 CR3 Evidence for Stepwise mechanism vs. Me3Si SiMe2Ph SiMe2Ph A B Me3Si SiMe2Ph *A is more stable than B by 38 kcal/mol * Me3Si SiMe2Ph Me3Si Jorgensen, JACS, 1986,107, 1496 Product ratios are equal from either starting material suggesting common intermediate cation Baran Lab Properties of Silicon Hafensteiner Evidence for Rapid Nucleophilic Attack Extraordinary Metallation Me SiMe3 SiMe3 Li Si t-BuLi Me SnCl4 Cl Me3Si Cl Me2Si Cl SiMe MeO OMe 3 OMe OMe Me2Si Cl vs. -

The Automated Flow Synthesis of Fluorine Containing Organic Compounds
The automated flow synthesis of fluorine containing organic compounds by CHANTAL SCHOLTZ Submitted in partial fulfilment of the requirements for the degree PHILOSOPHAE DOCTOR In the Faculty of Natural & Agricultural Sciences UNIVERSITY OF PRETORIA PRETORIA Supervisor: Dr D.L. Riley February 2019 DECLARATION I, Chantal Scholtz declare that the thesis/dissertation, which I hereby submit for the degree PhD Chemistry at the University of Pretoria, is my own work and has not previously been submitted by me for a degree at this or any other tertiary institution. Signature :.......................................... Date :..................................... ii ACKNOWLEDGEMENTS I would herewith sincerely like to show my gratitude to the following individuals for their help, guidance and assistance throughout the duration of this project: My supervisor, Doctor Darren Riley, for his knowledge and commitment. Thank you for being a fantastic supervisor and allowing me the opportunity to learn so many valuable skills. My husband, Clinton, for all your love, support and patience and for always being there for me. You are the best. My family, for all the encouragement and support you gave me as well as always believing in me. I will always appreciate what you have done for me. Mr Drikus van der Westhuizen and Mr Johan Postma for their assistance at the Pelchem laboratories with product isolation and characterisation. Dr Mamoalosi Selepe for NMR spectroscopy services, Jeanette Strydom for XRF services and Gerda Ehlers at the UP library for her invaluable assistance. All my friends and colleagues for the continuous moral support, numerous helpful discussions and necessary coffee breaks. My colleagues at Chemical Process Technologies for their ongoing support and motivation, especially Dr Hannes Malan and Prof. -

Syllabus CHEM 6352 2014
CHEM 6352 Organic Reactions & Synthesis Fall 2014 Jeremy A. May Office: 5025 SERC Office hours: T/Th 10-11 am or by appointment (email me) Email: [email protected] Website: http://mynsm.uh.edu/groups/maygroup/wiki/b24dc/Classes.html Lectures: 154 Fleming Tuesdays and Thursdays 8:30–10:00. August 26–December 6, 2014. Homework Session Saturdays 3:00 pm to 5:30 pm in Fleming 154/160/162. No class November 27–29, 2014 (Thanksgiving recess); Oct. 31st is last day to withdraw Optional Texts (on reserve at MD Anderson Library) Zweifel, G.; Nantz, M. “Modern Organic Synthesis: An Introduction” March, J. “Advanced Organic Chemistry” Corey, E. J.; Cheng, X.-M. “The Logic of Chemical Synthesis” Warren, S. “Designing Organic Syntheses: A Programmed Introduction to the Synthon Approach” Kürti, L.; Czakó, B. “Strategic Applications of Named Reactions in Organic Synthesis” Grossman, R. “The Art of Writing Reasonable Organic Reaction Mechanisms” Model Sets: Students are strongly encouraged to purchase at least one set. HGS biochemistry molecular model sets are recommended and are available at Research Stores in the Old Science Building. Other relevant texts and references: Greene; Wuts. “Protective Groups in Organic Synthesis” Nicolaou, K.C.; Sorensen, E. “Classics in Total Synthesis” Nicolaou, K.C.; Snyder, S. “Classics in Total Synthesis II” Larock, R. C. "Comprehensive Organic Transformations" Hartwig, J. “Organotransition Metal Chemistry: From Bonding to Catalysis” Tsuji, J. “Palladium Reagents and Catalysts” Hegedus, L. “Transition Metals in the Synthesis of Complex Organic Molecules” Problem Sets: Problem Sets will be distributed on Tuesdays (or before) and are due by the next Saturday at the Homework Session.