Nrf1 Is Endowed with a Dominant Tumor-Repressing Effect
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Luzhou Laojiao Co., Ltd. 2018 Annual Report April 2019
2018 Annual Report of Luzhou Laojiao Co.,Ltd. Luzhou Laojiao Co., Ltd. 2018 Annual Report April 2019 1 2018 Annual Report of Luzhou Laojiao Co.,Ltd. Section I Important Statements, Contents and Definitions The Board of Directors, Board of Supervisors, directors, supervisors and senior management guarantee that the information presented in this report is free of any false records, misleading statements or material omissions, and shall Individually and together be legally liable for truthfulness, accuracy and completeness of its contents. Mr. Liu Miao, responsible person for the Company, Ms. Xie Hong, responsible person for accounting work and Mr. Yan Li, responsible person for the Company’s financial affairs (Accounting Supervisor) have warranted that the financial statements in this report are true and complete. Other directors attended the board meeting to deliberate this report by themselves except the following directors. Name of directors who did not Position of directors who did not Reason for not attending Name of attend the meeting in person attend the meeting in person the meeting in person deputies Liu Miao Chairman of the Board work Lin Feng Shen Caihong Director work Jiang Yuhui Affected by risks, uncertainties and assumptions, the forward-looking statements concerning business objectives and future plans made in this report based on the subjective assumptions and judgments of the future policies and economic conditions may be significantly different from the actual results. Such statements shall not be considered as virtual promises of the Company to investors, and the investors and relevant persons shall maintain adequate risk awareness and shall understand the differences between plans, forecasts and commitments. -

Lanzhou-Chongqing Railway Development – Resettlement Action Plan Monitoring Report No
Resettlement Monitoring Report Project Number: 35354 April 2010 PRC: Lanzhou-Chongqing Railway Development – Resettlement Action Plan Monitoring Report No. 1 Prepared by: CIECC Overseas Consulting Co., Ltd Beijing, PRC For: Ministry of Railways This report has been submitted to ADB by the Ministry of Railways and is made publicly available in accordance with ADB’s public communications policy (2005). It does not necessarily reflect the views of ADB. The People’s Republic of China ADB Loan Lanzhou—Chongqing RAILWAY PROJECT EXTERNAL MONITORING & EVALUATION OF RESETTLEMENT ACTION PLAN Report No.1 Prepared by CIECC OVERSEAS CONSULTING CO.,LTD April 2010 Beijing 10 ADB LOAN EXTERNAL Monitoring Report– No. 1 TABLE OF CONTENTS PREFACE 4 OVERVIEW..................................................................................................................................................... 5 1. PROJECT BRIEF DESCRIPTION .......................................................................................................................7 2. PROJECT AND RESETTLEMENT PROGRESS ................................................................................................10 2.1 PROJECT PROGRESS ...............................................................................................................................10 2.2 LAND ACQUISITION, HOUSE DEMOLITION AND RESETTLEMENT PROGRESS..................................................10 3. MONITORING AND EVALUATION .................................................................................................................14 -
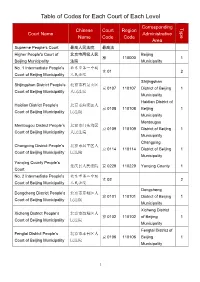
Table of Codes for Each Court of Each Level
Table of Codes for Each Court of Each Level Corresponding Type Chinese Court Region Court Name Administrative Name Code Code Area Supreme People’s Court 最高人民法院 最高法 Higher People's Court of 北京市高级人民 Beijing 京 110000 1 Beijing Municipality 法院 Municipality No. 1 Intermediate People's 北京市第一中级 京 01 2 Court of Beijing Municipality 人民法院 Shijingshan Shijingshan District People’s 北京市石景山区 京 0107 110107 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Haidian District of Haidian District People’s 北京市海淀区人 京 0108 110108 Beijing 1 Court of Beijing Municipality 民法院 Municipality Mentougou Mentougou District People’s 北京市门头沟区 京 0109 110109 District of Beijing 1 Court of Beijing Municipality 人民法院 Municipality Changping Changping District People’s 北京市昌平区人 京 0114 110114 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Yanqing County People’s 延庆县人民法院 京 0229 110229 Yanqing County 1 Court No. 2 Intermediate People's 北京市第二中级 京 02 2 Court of Beijing Municipality 人民法院 Dongcheng Dongcheng District People’s 北京市东城区人 京 0101 110101 District of Beijing 1 Court of Beijing Municipality 民法院 Municipality Xicheng District Xicheng District People’s 北京市西城区人 京 0102 110102 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Fengtai District of Fengtai District People’s 北京市丰台区人 京 0106 110106 Beijing 1 Court of Beijing Municipality 民法院 Municipality 1 Fangshan District Fangshan District People’s 北京市房山区人 京 0111 110111 of Beijing 1 Court of Beijing Municipality 民法院 Municipality Daxing District of Daxing District People’s 北京市大兴区人 京 0115 -

CHINA VANKE CO., LTD.* 萬科企業股份有限公司 (A Joint Stock Company Incorporated in the People’S Republic of China with Limited Liability) (Stock Code: 2202)
Hong Kong Exchanges and Clearing Limited and The Stock Exchange of Hong Kong Limited take no responsibility for the contents of this announcement, make no representation as to its accuracy or completeness and expressly disclaim any liability whatsoever for any loss howsoever arising from or in reliance upon the whole or any part of the contents of this announcement. CHINA VANKE CO., LTD.* 萬科企業股份有限公司 (A joint stock company incorporated in the People’s Republic of China with limited liability) (Stock Code: 2202) 2019 ANNUAL RESULTS ANNOUNCEMENT The board of directors (the “Board”) of China Vanke Co., Ltd.* (the “Company”) is pleased to announce the audited results of the Company and its subsidiaries for the year ended 31 December 2019. This announcement, containing the full text of the 2019 Annual Report of the Company, complies with the relevant requirements of the Rules Governing the Listing of Securities on The Stock Exchange of Hong Kong Limited in relation to information to accompany preliminary announcement of annual results. Printed version of the Company’s 2019 Annual Report will be delivered to the H-Share Holders of the Company and available for viewing on the websites of The Stock Exchange of Hong Kong Limited (www.hkexnews.hk) and of the Company (www.vanke.com) in April 2020. Both the Chinese and English versions of this results announcement are available on the websites of the Company (www.vanke.com) and The Stock Exchange of Hong Kong Limited (www.hkexnews.hk). In the event of any discrepancies in interpretations between the English version and Chinese version, the Chinese version shall prevail, except for the financial report prepared in accordance with International Financial Reporting Standards, of which the English version shall prevail. -
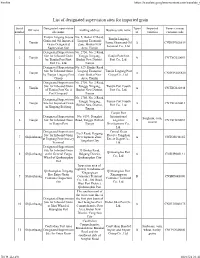
List of Designated Supervision Sites for Imported Grain
Firefox https://translate.googleusercontent.com/translate_f List of designated supervision sites for imported grain Serial Designated supervision Types Imported Venue (venue) Off zone mailing address Business unit name number site name of varieties customs code Tianjin Lingang Jiayue No. 5, Bohai 37 Road, Tianjin Lingang Grain and Oil Imported Lingang Economic 1 Tianjin Jiayue Grain and Oil A CNDGN02S619 Grain Designated Zone, Binhai New Terminal Co., Ltd. Supervision Site Area, Tianjin Designated Supervision No. 2750, No. 2 Road, Site for Inbound Grain Tanggu Xingang, Tianjin Port First 2 Tianjin A CNTXG020051 by Tianjin Port First Binhai New District, Port Co., Ltd. Port Co., Ltd. Tianjin Designated Supervision No. 529, Hunhe Road, Site for Inbound Grain Lingang Economic Tianjin Lingang Port 3 Tianjin A CNDGN02S620 by Tianjin Lingang Port Zone, Binhai New Group Co., Ltd. Group Area, Tianjin Designated Supervision No. 2750, No. 2 Road, Site for Inbound Grain Tanggu Xingang, Tianjin Port Fourth 4 Tianjin A CNTXG020448 of Tianjin Port No. 4 Binhai New District, Port Co., Ltd. Port Company Tianjin No. 2750, No. 2 Road, Designated Supervision Tanggu Xingang, Tianjin Port Fourth 5 Tianjin Site for Imported Grain A CNTXG020413 Binhai New District, Port Co., Ltd. in Xingang Beijiang Tianjin Tianjin Port Designated Supervision No. 6199, Donghai International Sorghum, corn, 6 Tianjin Site for Inbound Grain Road, Tanggu District, Logistics B CNTXG020051 sesame in Tianjin Port Tianjin Development Co., Ltd. Designated Supervision Central Grain No.9 Road, Haigang Site for Inbound Grain Reserve Tangshan 7 Shijiazhuang Development Zone, A CNTGS040165 at Jingtang Port Grocery Direct Depot Co., Tangshan City Terminal Ltd. -

Supplemental Information Nrf1 Is Endowed with a Dominant
Supplemental information Nrf1 is endowed with a dominant tumor-repressing effect onto the Wnt/-Catenin -dependent and -independent signaling networks in the human liver cancer Jiayu Chen1,2,§, Meng Wang1,§, Xufang Ru1,3,§, Yuancai Xiang1,4,§, Yonggang Ren1,5,§, Xiping Liu2, Lu Qiu1,6, and Yiguo Zhang1* 1The Laboratory of Cell Biochemistry and Topogenetic Regulation, College of Bioengineering and Faculty of Sciences, Chongqing University, No. 174 Shazheng Street, Shapingba District, Chongqing 400044, China. 2Department of Biochemistry and Molecular Biology, Zunyi Medical University, No. 6 Xuefu-Xi Road, Xinpu New District, Zunyi 563000, Guizhou, China. 3Department of Neurosurgery, Southwest Hospital, Army (Third Military) Medical University, No. 29 Gaotanyan Street, Shapingba District, Chongqing 400038, China. 4Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Southwest Medical University, No. 1 at the First Section of Xianglin Road, Longmatan District, Luzhou 646000, Sichuan, China. 5Department of Biochemistry, North Sichuan Medical College, No. 55 Dongshun Road, Gaoping District, Nanchong 637000, Sichuan, China. 6School of Life Sciences, Zhengzhou University, No. 100 Kexue Avenue, Zhengzhou 450001, Henan, China. §Contributed equally to this work. *Correspondence should be addressed to Yiguo Zhang (Email: [email protected], or [email protected]) 1. Supplemental materials and methods 1.1 The TOPflash Luciferase reporter assay to measure β-catenin/Tcf-driven transcriptional activity. Human 293T cells (2×104) were seeded in each well of a 48-vial plate and allowed for growth to 70% confluence. The cells were co-transfected with 100 ng of the firefly luciferase reporter called TOPflash (driven by the consensus β-catenin/Tcf4-binding site) or its mutant control plasmid called FOPflash, together with 5 ng of Renilla luciferase reporter (pRL-CMV), plus 10 pmol of indicated small interference RNA targeting for Nrf1 (i.e., siNrf1) or a scrambled negative control RNA (i.e. -

Luzhou Laojiao Co., Ltd. 2019 Annual Report April 2020
2019 Annual Report of Luzhou Laojiao Co., Ltd. Luzhou Laojiao Co., Ltd. 2019 Annual Report April 2020 1 2019 Annual Report of Luzhou Laojiao Co., Ltd. Section I Important Statements, Contents and Definitions The Board of Directors, Board of Supervisors, directors, supervisors and senior management guarantee that the information presented in this report is free of any false records, misleading statements or material omissions, and shall individually and together be legally liable for truthfulness, accuracy and completeness of its contents. Mr. Liu Miao, responsible person for the Company, Ms. Xie Hong, responsible person for accounting and Ms. Yan Li, responsible person for the Company’s financial affairs (Accounting Supervisor) have warranted that the financial statements in this report are true and complete. Other directors attended the board meeting to deliberate this report by themselves except the following directors. Position of directors who Name of directors who did not Reason for not attending did not attend the meeting Name of deputies attend the meeting in person the meeting in person in person Lin Feng Director Work Liu Miao Affected by risks, uncertainties and assumptions, the forward-looking statements concerning business objectives and future plans made in this report based on the subjective assumptions and judgments of the future policies and economic conditions may be significantly different from the actual results. Such statements shall not be considered as virtual promises of the Company to investors, and the investors and relevant persons shall maintain adequate risk awareness and shall understand the differences between plans, forecasts and commitments. In the annual report, the potential risks in the operation of the Company have been disclosed. -

Hepg2) Cells Via Wnt/Β‑Catenin Pathway Yonggang Ren1,2*, Yujiao Wang3*, Shuai Hao3, Yuhan Yang3, Wendong Xiong3, Lu Qiu4, Jia Tao5, Aifa Tang1
Journal of Cancer 2020, Vol. 11 6939 Ivyspring International Publisher Journal of Cancer 2020; 11(23): 6939-6949. doi: 10.7150/jca.48100 Research Paper NFE2L3 promotes malignant behavior and EMT of human hepatocellular carcinoma (HepG2) cells via Wnt/β‑catenin pathway Yonggang Ren1,2*, Yujiao Wang3*, Shuai Hao3, Yuhan Yang3, Wendong Xiong3, Lu Qiu4, Jia Tao5, Aifa Tang1 1. Guangdong Key Laboratory of Systems Biology and Synthetic Biology for Urogenital Tumors, Institute of Translational Medicine, First Affiliated Hospital of Shenzhen University, Shenzhen Second People’s Hospital, Shenzhen, Guangdong 518035, P.R. China. 2. Department of Biochemistry, North Sichuan Medical College, Nanchong, Sichuan 637000, P.R. China. 3. Department of Clinical Medicine, North Sichuan Medical College, Nanchong, Sichuan 637000, P.R. China. 4. School of Life Sciences, Zhengzhou University, Zhengzhou 450001, P.R. China. 5. Department of Pathology, Affiliated Hospital of North Sichuan Medical College, Nanchong, Sichuan 637000, P.R. China. *These authors contributed equally to this work. Corresponding authors: Aifa Tang, Guangdong Key Laboratory of Systems Biology and Synthetic Biology for Urogenital Tumors, Institute of Translational Medicine, First Affiliated Hospital of Shenzhen University, Shenzhen Second People’s Hospital. No. 3002 Sungang Xi Road, Futian District, Shenzhen, Guangdong, 518035, P.R. China; Tel: (86) 755-8300-3435; Fax: (86) 755-8300-3435; E-mail: [email protected]; Yonggang Ren, Guangdong Key Laboratory of Systems Biology and Synthetic Biology for Urogenital Tumors, Institute of Translational Medicine, First Affiliated Hospital of Shenzhen University, Shenzhen Second People’s Hospital. No. 3002 Sungang Xi Road, Futian District, Shenzhen, Guangdong, 518035, P.R. -

Exploring the Spatial Pattern of Urban Block Development Based on POI Analysis: a Case Study in Wuhan, China
sustainability Article Exploring the Spatial Pattern of Urban Block Development Based on POI Analysis: A Case Study in Wuhan, China Hailing Xu, Jianghong Zhu * and Zhanqi Wang School of Public Administration, China University of Geosciences, Wuhan 430074, China; [email protected] (H.X.); [email protected] (Z.W.) * Correspondence: [email protected]; Tel.: +86-1339-715-7676 Received: 3 November 2019; Accepted: 4 December 2019; Published: 6 December 2019 Abstract: As a kind of geospatial big data, point of interest (POI) with useful information has become a hot research topic. Compared with traditional methods, big data has great potential in developing a more accurate method for identifying the urban spatial pattern. This paper uses diversity index and kernel density analysis of POI data on several types of urban infrastructure to investigate the identification of urban block development stages in Wuhan, and divides them into the primary, growth, and mature stage. Its accuracy is verified by exploring urban micro-centers. Results show that: (1) the spatial pattern of urban blocks in Wuhan presents the distribution of “mature blocks concentrated like a core, growth blocks distributed like two wings, and primary blocks with wide range distributed surround”; (2) areas with more connected construction land and streets with better socio-economic status tend to have a higher level of maturity, vice versa; (3) balancing the number of micro-centers at different stages is beneficial to promote the flattened urban development of Wuhan in the future. The research proves that this method is feasible, and it is also applicable to the study of urban spatial pattern in other cities. -

CHENGDU-NANCHONG EXPRESSWAY PROJECT (Loan 1638-PRC)
ASIAN DEVELOPMENT BANK PCR: PRC 30082 PROJECT COMPLETION REPORT ON THE CHENGDU-NANCHONG EXPRESSWAY PROJECT (Loan 1638-PRC) IN THE PEOPLE’S REPUBLIC OF CHINA December 2004 CURRENCY EQUIVALENTS Currency Unit – yuan (CNY) At Appraisal At Project Completion (19 July 1998) (20 August 2004) CNY1.00 = $0.1208 $0.1208 $1.00 = CNY8.2768 CNY8.2768 ABBREVIATIONS AADT – annual average daily traffic ADB – Asian Development Bank BME – benefit monitoring and evaluation EIA – environmental impact assessment EIRR – economic internal rate of return FIRR – financial internal rate of return GDP – gross domestic product ha – hectare ICB – international competitive bidding km – kilometer LCB – local competitive bidding LIBOR – London interbank offered rate MTE – medium truck equivalent MOC – Ministry of Communications NTHS – national trunk highway system O&M – operation and maintenance PCR – project completion report PRC – The People’s Republic of China PRCM – Asian Development Bank Resident Mission in the People’s Republic of China SCELLC – Sichuan Chengnan Expressway Limited Liability Company SPCD – Sichuan Provincial Communications Department SPG – Sichuan Provincial Government TA – technical assistance VOC – vehicle operating cost WACC – weighted average cost of capital NOTE In this report, "$" refers to US dollars. CONTENTS Page BASIC DATA ii MAPS vii I. PROJECT DESCRIPTION 1 II. EVALUATION OF DESIGN AND IMPLEMENTATION 2 A. Relevance of Design and Formulation 2 B. Project Outputs 2 C. Project Costs and Financing Plan 4 D. Disbursements 5 E. Project Schedule 5 F. Implementation Arrangements 6 G. Conditions and Covenants 6 H. Consultant Recruitment and Procurement 6 I. Performance of Consultants, Contractors, and Suppliers 7 J. Performance of the Borrower and the Executing Agency 7 K. -

Global Offering
瀘州市商業銀行股份有限公司 Luzhou City Commercial Bank Co., Ltd. (A joint stock company incorporated in the People's Republic of China with limited liability) Stock Code: 1983 Global offerinG Sole Sponsor Joint Global Coordinators, Joint Bookrunners and Joint Lead Managers Joint Lead Managers IMPORTANT IMPORTANT: If you are in doubt about any information contained in this prospectus you should obtain independent professional advice. Luzhou City Commercial Bank Co., Ltd.* 瀘州市商業銀行股份有限公司* (A joint stock company incorporated in the People’s Republic of China with limited liability) GLOBAL OFFERING Number of Offer Shares in the : 545,740,000 H Shares (subject to the Global Offering Over-allotment Option) Number of International Offer Shares : 491,166,000 H Shares (subject to adjustment and the Over-allotment Option) Number of Hong Kong Offer Shares : 54,574,000 H Shares (subject to adjustment) Maximum Offer Price : HK$3.40 per H Share, plus brokerage of 1%, SFC transaction levy of 0.0027% and Hong Kong Stock Exchange trading fee of 0.005% (payable in full on application in Hong Kong dollars, subject to refund) Nominal value : RMB1.00 per H Share Stock code : 1983 Sole Sponsor Joint Global Coordinators, Joint Bookrunners and Joint Lead Managers Joint Lead Managers Hong Kong Exchanges and Clearing Limited, The Stock Exchange of Hong Kong Limited and Hong Kong Securities Clearing Company Limited take no responsibility for the contents of this prospectus, make no representation as to its accuracy or completeness and expressly disclaim any liability whatsoever for any loss howsoever arising from or in reliance upon the whole or any part of the contents of this prospectus. -

Dec 13, 2018 Prospectus
IMPORTANT If you are in any doubt about any of the contents of this prospectus, you should obtain independent professional advice. 四川能投發展股份有限公司 Sichuan Energy Investment Development Co., Ltd.* (a joint stock company incorporated in the People’s Republic of China with limited liability) GLOBAL OFFERING Number of Offer Shares under : 268,800,000 H Shares (subject to the the Global Offering Over-allotment Option) Number of Hong Kong Offer Shares : 26,880,000 H Shares (subject to adjustment) Number of International Offer Shares : 241,920,000 H Shares (subject to adjustment and the Over-allotment Option) Maximum Offer Price : HK$2.34 per H Share, plus brokerage of 1.0%, SFC transaction levy of 0.0027% and a Stock Exchange trading fee of 0.005% (payable in full on application in Hong Kong dollars and subject to refund) Nominal value : RMB1.00 per H Share Stock code : 1713 Sole Sponsor and Sole Global Coordinator Joint Bookrunners and Joint Lead Managers Hong Kong Exchanges and Clearing Limited, The Stock Exchange of Hong Kong Limited and Hong Kong Securities Clearing Company Limited take no responsibility for the contents of this prospectus, make no representation as to its accuracy or completeness and expressly disclaim any liability whatsoever for any loss howsoever arising from or in reliance upon the whole or any part of the contents of this prospectus. A copy of this prospectus, having attached thereto the documents specified in the paragraph headed “Documents Delivered to the Registrar of Companies and Available for Inspection” in Appendix VII to this prospectus, has been registered by the Registrar of Companies in Hong Kong as required by Section 342C of the Companies (Winding Up and Miscellaneous Provisions) Ordinance, Chapter 32 of the Laws of Hong Kong.