Lec 1 Thermodynamic Fuctions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Thermodynamic Potentials and Natural Variables
Revista Brasileira de Ensino de Física, vol. 42, e20190127 (2020) Articles www.scielo.br/rbef cb DOI: http://dx.doi.org/10.1590/1806-9126-RBEF-2019-0127 Licença Creative Commons Thermodynamic Potentials and Natural Variables M. Amaku1,2, F. A. B. Coutinho*1, L. N. Oliveira3 1Universidade de São Paulo, Faculdade de Medicina, São Paulo, SP, Brasil 2Universidade de São Paulo, Faculdade de Medicina Veterinária e Zootecnia, São Paulo, SP, Brasil 3Universidade de São Paulo, Instituto de Física de São Carlos, São Carlos, SP, Brasil Received on May 30, 2019. Revised on September 13, 2018. Accepted on October 4, 2019. Most books on Thermodynamics explain what thermodynamic potentials are and how conveniently they describe the properties of physical systems. Certain books add that, to be useful, the thermodynamic potentials must be expressed in their “natural variables”. Here we show that, given a set of physical variables, an appropriate thermodynamic potential can always be defined, which contains all the thermodynamic information about the system. We adopt various perspectives to discuss this point, which to the best of our knowledge has not been clearly presented in the literature. Keywords: Thermodynamic Potentials, natural variables, Legendre transforms. 1. Introduction same statement cannot be applied to the temperature. In real fluids, even in simple ones, the proportionality Basic concepts are most easily understood when we dis- to T is washed out, and the Internal Energy is more cuss simple systems. Consider an ideal gas in a cylinder. conveniently expressed as a function of the entropy and The cylinder is closed, its walls are conducting, and a volume: U = U(S, V ). -

Lab: Specific Heat
Lab: Specific Heat Summary Students relate thermal energy to heat capacity by comparing the heat capacities of different materials and graphing the change in temperature over time for a specific material. Students explore the idea of insulators and conductors in an attempt to apply their knowledge in real world applications, such as those that engineers would. Engineering Connection Engineers must understand the heat capacity of insulators and conductors if they wish to stay competitive in “green” enterprises. Engineers who design passive solar heating systems take advantage of the natural heat capacity characteristics of materials. In areas of cooler climates engineers chose building materials with a low heat capacity such as slate roofs in an attempt to heat the home during the day. In areas of warmer climates tile roofs are popular for their ability to store the sun's heat energy for an extended time and release it slowly after the sun sets, to prevent rapid temperature fluctuations. Engineers also consider heat capacity and thermal energy when they design food containers and household appliances. Just think about your aluminum soda can versus glass bottles! Grade Level: 9-12 Group Size: 4 Time Required: 90 minutes Activity Dependency :None Expendable Cost Per Group : US$ 5 The cost is less if thermometers are available for each group. NYS Science Standards: STANDARD 1 Analysis, Inquiry, and Design STANDARD 4 Performance indicator 2.2 Keywords: conductor, energy, energy storage, heat, specific heat capacity, insulator, thermal energy, thermometer, thermocouple Learning Objectives After this activity, students should be able to: Define heat capacity. Explain how heat capacity determines a material's ability to store thermal energy. -

PDF Version of Helmholtz Free Energy
Free energy Free Energy at Constant T and V Starting with the First Law dU = δw + δq At constant temperature and volume we have δw = 0 and dU = δq Free Energy at Constant T and V Starting with the First Law dU = δw + δq At constant temperature and volume we have δw = 0 and dU = δq Recall that dS ≥ δq/T so we have dU ≤ TdS Free Energy at Constant T and V Starting with the First Law dU = δw + δq At constant temperature and volume we have δw = 0 and dU = δq Recall that dS ≥ δq/T so we have dU ≤ TdS which leads to dU - TdS ≤ 0 Since T and V are constant we can write this as d(U - TS) ≤ 0 The quantity in parentheses is a measure of the spontaneity of the system that depends on known state functions. Definition of Helmholtz Free Energy We define a new state function: A = U -TS such that dA ≤ 0. We call A the Helmholtz free energy. At constant T and V the Helmholtz free energy will decrease until all possible spontaneous processes have occurred. At that point the system will be in equilibrium. The condition for equilibrium is dA = 0. A time Definition of Helmholtz Free Energy Expressing the change in the Helmholtz free energy we have ∆A = ∆U – T∆S for an isothermal change from one state to another. The condition for spontaneous change is that ∆A is less than zero and the condition for equilibrium is that ∆A = 0. We write ∆A = ∆U – T∆S ≤ 0 (at constant T and V) Definition of Helmholtz Free Energy Expressing the change in the Helmholtz free energy we have ∆A = ∆U – T∆S for an isothermal change from one state to another. -

Chapter 3. Second and Third Law of Thermodynamics
Chapter 3. Second and third law of thermodynamics Important Concepts Review Entropy; Gibbs Free Energy • Entropy (S) – definitions Law of Corresponding States (ch 1 notes) • Entropy changes in reversible and Reduced pressure, temperatures, volumes irreversible processes • Entropy of mixing of ideal gases • 2nd law of thermodynamics • 3rd law of thermodynamics Math • Free energy Numerical integration by computer • Maxwell relations (Trapezoidal integration • Dependence of free energy on P, V, T https://en.wikipedia.org/wiki/Trapezoidal_rule) • Thermodynamic functions of mixtures Properties of partial differential equations • Partial molar quantities and chemical Rules for inequalities potential Major Concept Review • Adiabats vs. isotherms p1V1 p2V2 • Sign convention for work and heat w done on c=C /R vm system, q supplied to system : + p1V1 p2V2 =Cp/CV w done by system, q removed from system : c c V1T1 V2T2 - • Joule-Thomson expansion (DH=0); • State variables depend on final & initial state; not Joule-Thomson coefficient, inversion path. temperature • Reversible change occurs in series of equilibrium V states T TT V P p • Adiabatic q = 0; Isothermal DT = 0 H CP • Equations of state for enthalpy, H and internal • Formation reaction; enthalpies of energy, U reaction, Hess’s Law; other changes D rxn H iD f Hi i T D rxn H Drxn Href DrxnCpdT Tref • Calorimetry Spontaneous and Nonspontaneous Changes First Law: when one form of energy is converted to another, the total energy in universe is conserved. • Does not give any other restriction on a process • But many processes have a natural direction Examples • gas expands into a vacuum; not the reverse • can burn paper; can't unburn paper • heat never flows spontaneously from cold to hot These changes are called nonspontaneous changes. -

Compression of an Ideal Gas with Temperature Dependent Specific
Session 3666 Compression of an Ideal Gas with Temperature-Dependent Specific Heat Capacities Donald W. Mueller, Jr., Hosni I. Abu-Mulaweh Engineering Department Indiana University–Purdue University Fort Wayne Fort Wayne, IN 46805-1499, USA Overview The compression of gas in a steady-state, steady-flow (SSSF) compressor is an important topic that is addressed in virtually all engineering thermodynamics courses. A typical situation encountered is when the gas inlet conditions, temperature and pressure, are known and the compressor dis- charge pressure is specified. The traditional instructional approach is to make certain assumptions about the compression process and about the gas itself. For example, a special case often consid- ered is that the compression process is reversible and adiabatic, i.e. isentropic. The gas is usually ÊÌ considered to be ideal, i.e. ÈÚ applies, with either constant or temperature-dependent spe- cific heat capacities. The constant specific heat capacity assumption allows for direct computation of the discharge temperature, while the temperature-dependent specific heat assumption does not. In this paper, a MATLAB computer model of the compression process is presented. The substance being compressed is air which is considered to be an ideal gas with temperature-dependent specific heat capacities. Two approaches are presented to describe the temperature-dependent properties of air. The first approach is to use the ideal gas table data from Ref. [1] with a look-up interpolation scheme. The second approach is to use a curve fit with the NASA Lewis coefficients [2,3]. The computer model developed makes use of a bracketing–bisection algorithm [4] to determine the temperature of the air after compression. -

Thermodynamics
ME346A Introduction to Statistical Mechanics { Wei Cai { Stanford University { Win 2011 Handout 6. Thermodynamics January 26, 2011 Contents 1 Laws of thermodynamics 2 1.1 The zeroth law . .3 1.2 The first law . .4 1.3 The second law . .5 1.3.1 Efficiency of Carnot engine . .5 1.3.2 Alternative statements of the second law . .7 1.4 The third law . .8 2 Mathematics of thermodynamics 9 2.1 Equation of state . .9 2.2 Gibbs-Duhem relation . 11 2.2.1 Homogeneous function . 11 2.2.2 Virial theorem / Euler theorem . 12 2.3 Maxwell relations . 13 2.4 Legendre transform . 15 2.5 Thermodynamic potentials . 16 3 Worked examples 21 3.1 Thermodynamic potentials and Maxwell's relation . 21 3.2 Properties of ideal gas . 24 3.3 Gas expansion . 28 4 Irreversible processes 32 4.1 Entropy and irreversibility . 32 4.2 Variational statement of second law . 32 1 In the 1st lecture, we will discuss the concepts of thermodynamics, namely its 4 laws. The most important concepts are the second law and the notion of Entropy. (reading assignment: Reif x 3.10, 3.11) In the 2nd lecture, We will discuss the mathematics of thermodynamics, i.e. the machinery to make quantitative predictions. We will deal with partial derivatives and Legendre transforms. (reading assignment: Reif x 4.1-4.7, 5.1-5.12) 1 Laws of thermodynamics Thermodynamics is a branch of science connected with the nature of heat and its conver- sion to mechanical, electrical and chemical energy. (The Webster pocket dictionary defines, Thermodynamics: physics of heat.) Historically, it grew out of efforts to construct more efficient heat engines | devices for ex- tracting useful work from expanding hot gases (http://www.answers.com/thermodynamics). -

Thermodynamic Analysis Based on the Second-Order Variations of Thermodynamic Potentials
Theoret. Appl. Mech., Vol.35, No.1-3, pp. 215{234, Belgrade 2008 Thermodynamic analysis based on the second-order variations of thermodynamic potentials Vlado A. Lubarda ¤ Abstract An analysis of the Gibbs conditions of stable thermodynamic equi- librium, based on the constrained minimization of the four fundamen- tal thermodynamic potentials, is presented with a particular attention given to the previously unexplored connections between the second- order variations of thermodynamic potentials. These connections are used to establish the convexity properties of all potentials in relation to each other, which systematically deliver thermodynamic relationships between the speci¯c heats, and the isentropic and isothermal bulk mod- uli and compressibilities. The comparison with the classical derivation is then given. Keywords: Gibbs conditions, internal energy, second-order variations, speci¯c heats, thermodynamic potentials 1 Introduction The Gibbs conditions of thermodynamic equilibrium are of great importance in the analysis of the equilibrium and stability of homogeneous and heterogeneous thermodynamic systems [1]. The system is in a thermodynamic equilibrium if its state variables do not spontaneously change with time. As a consequence of the second law of thermodynamics, the equilibrium state of an isolated sys- tem at constant volume and internal energy is the state with the maximum value of the total entropy. Alternatively, among all neighboring states with the ¤Department of Mechanical and Aerospace Engineering, University of California, San Diego; La Jolla, CA 92093-0411, USA and Montenegrin Academy of Sciences and Arts, Rista Stijovi¶ca5, 81000 Podgorica, Montenegro, e-mail: [email protected]; [email protected] 215 216 Vlado A. Lubarda same volume and total entropy, the equilibrium state is one with the lowest total internal energy. -

Chapter 3 3.4-2 the Compressibility Factor Equation of State
Chapter 3 3.4-2 The Compressibility Factor Equation of State The dimensionless compressibility factor, Z, for a gaseous species is defined as the ratio pv Z = (3.4-1) RT If the gas behaves ideally Z = 1. The extent to which Z differs from 1 is a measure of the extent to which the gas is behaving nonideally. The compressibility can be determined from experimental data where Z is plotted versus a dimensionless reduced pressure pR and reduced temperature TR, defined as pR = p/pc and TR = T/Tc In these expressions, pc and Tc denote the critical pressure and temperature, respectively. A generalized compressibility chart of the form Z = f(pR, TR) is shown in Figure 3.4-1 for 10 different gases. The solid lines represent the best curves fitted to the data. Figure 3.4-1 Generalized compressibility chart for various gases10. It can be seen from Figure 3.4-1 that the value of Z tends to unity for all temperatures as pressure approach zero and Z also approaches unity for all pressure at very high temperature. If the p, v, and T data are available in table format or computer software then you should not use the generalized compressibility chart to evaluate p, v, and T since using Z is just another approximation to the real data. 10 Moran, M. J. and Shapiro H. N., Fundamentals of Engineering Thermodynamics, Wiley, 2008, pg. 112 3-19 Example 3.4-2 ---------------------------------------------------------------------------------- A closed, rigid tank filled with water vapor, initially at 20 MPa, 520oC, is cooled until its temperature reaches 400oC. -
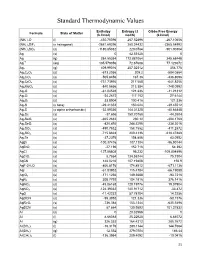
Standard Thermodynamic Values
Standard Thermodynamic Values Enthalpy Entropy (J Gibbs Free Energy Formula State of Matter (kJ/mol) mol/K) (kJ/mol) (NH4)2O (l) -430.70096 267.52496 -267.10656 (NH4)2SiF6 (s hexagonal) -2681.69296 280.24432 -2365.54992 (NH4)2SO4 (s) -1180.85032 220.0784 -901.90304 Ag (s) 0 42.55128 0 Ag (g) 284.55384 172.887064 245.68448 Ag+1 (aq) 105.579056 72.67608 77.123672 Ag2 (g) 409.99016 257.02312 358.778 Ag2C2O4 (s) -673.2056 209.2 -584.0864 Ag2CO3 (s) -505.8456 167.36 -436.8096 Ag2CrO4 (s) -731.73976 217.568 -641.8256 Ag2MoO4 (s) -840.5656 213.384 -748.0992 Ag2O (s) -31.04528 121.336 -11.21312 Ag2O2 (s) -24.2672 117.152 27.6144 Ag2O3 (s) 33.8904 100.416 121.336 Ag2S (s beta) -29.41352 150.624 -39.45512 Ag2S (s alpha orthorhombic) -32.59336 144.01328 -40.66848 Ag2Se (s) -37.656 150.70768 -44.3504 Ag2SeO3 (s) -365.2632 230.12 -304.1768 Ag2SeO4 (s) -420.492 248.5296 -334.3016 Ag2SO3 (s) -490.7832 158.1552 -411.2872 Ag2SO4 (s) -715.8824 200.4136 -618.47888 Ag2Te (s) -37.2376 154.808 43.0952 AgBr (s) -100.37416 107.1104 -96.90144 AgBrO3 (s) -27.196 152.716 54.392 AgCl (s) -127.06808 96.232 -109.804896 AgClO2 (s) 8.7864 134.55744 75.7304 AgCN (s) 146.0216 107.19408 156.9 AgF•2H2O (s) -800.8176 174.8912 -671.1136 AgI (s) -61.83952 115.4784 -66.19088 AgIO3 (s) -171.1256 149.3688 -93.7216 AgN3 (s) 308.7792 104.1816 376.1416 AgNO2 (s) -45.06168 128.19776 19.07904 AgNO3 (s) -124.39032 140.91712 -33.472 AgO (s) -11.42232 57.78104 14.2256 AgOCN (s) -95.3952 121.336 -58.1576 AgReO4 (s) -736.384 153.1344 -635.5496 AgSCN (s) 87.864 130.9592 101.37832 Al (s) -

Entropy Determination of Single-Phase High Entropy Alloys with Different Crystal Structures Over a Wide Temperature Range
entropy Article Entropy Determination of Single-Phase High Entropy Alloys with Different Crystal Structures over a Wide Temperature Range Sebastian Haas 1, Mike Mosbacher 1, Oleg N. Senkov 2 ID , Michael Feuerbacher 3, Jens Freudenberger 4,5 ID , Senol Gezgin 6, Rainer Völkl 1 and Uwe Glatzel 1,* ID 1 Metals and Alloys, University Bayreuth, 95447 Bayreuth, Germany; [email protected] (S.H.); [email protected] (M.M.); [email protected] (R.V.) 2 UES, Inc., 4401 Dayton-Xenia Rd., Dayton, OH 45432, USA; [email protected] 3 Institut für Mikrostrukturforschung, Forschungszentrum Jülich, 52425 Jülich, Germany; [email protected] 4 Leibniz Institute for Solid State and Materials Research Dresden (IFW Dresden), 01069 Dresden, Germany; [email protected] 5 TU Bergakademie Freiberg, Institut für Werkstoffwissenschaft, 09599 Freiberg, Germany 6 NETZSCH Group, Analyzing & Testing, 95100 Selb, Germany; [email protected] * Correspondence: [email protected]; Tel.: +49-921-55-5555 Received: 31 July 2018; Accepted: 20 August 2018; Published: 30 August 2018 Abstract: We determined the entropy of high entropy alloys by investigating single-crystalline nickel and five high entropy alloys: two fcc-alloys, two bcc-alloys and one hcp-alloy. Since the configurational entropy of these single-phase alloys differs from alloys using a base element, it is important to quantify ◦ the entropy. Using differential scanning calorimetry, cp-measurements are carried out from −170 C to the materials’ solidus temperatures TS. From these experiments, we determined the thermal entropy and compared it to the configurational entropy for each of the studied alloys. -

Pressure Vs. Volume and Boyle's
Pressure vs. Volume and Boyle’s Law SCIENTIFIC Boyle’s Law Introduction In 1642 Evangelista Torricelli, who had worked as an assistant to Galileo, conducted a famous experiment demonstrating that the weight of air would support a column of mercury about 30 inches high in an inverted tube. Torricelli’s experiment provided the first measurement of the invisible pressure of air. Robert Boyle, a “skeptical chemist” working in England, was inspired by Torricelli’s experiment to measure the pressure of air when it was compressed or expanded. The results of Boyle’s experiments were published in 1662 and became essentially the first gas law—a mathematical equation describing the relationship between the volume and pressure of air. What is Boyle’s law and how can it be demonstrated? Concepts • Gas properties • Pressure • Boyle’s law • Kinetic-molecular theory Background Open end Robert Boyle built a simple apparatus to measure the relationship between the pressure and volume of air. The apparatus ∆h ∆h = 29.9 in. Hg consisted of a J-shaped glass tube that was Sealed end 1 sealed at one end and open to the atmosphere V2 = /2V1 Trapped air (V1) at the other end. A sample of air was trapped in the sealed end by pouring mercury into Mercury the tube (see Figure 1). In the beginning of (Hg) the experiment, the height of the mercury Figure 1. Figure 2. column was equal in the two sides of the tube. The pressure of the air trapped in the sealed end was equal to that of the surrounding air and equivalent to 29.9 inches (760 mm) of mercury. -

The Application of Temperature And/Or Pressure Correction Factors in Gas Measurement COMBINED BOYLE’S – CHARLES’ GAS LAWS
The Application of Temperature and/or Pressure Correction Factors in Gas Measurement COMBINED BOYLE’S – CHARLES’ GAS LAWS To convert measured volume at metered pressure and temperature to selling volume at agreed base P1 V1 P2 V2 pressure and temperature = T1 T2 Temperatures & Pressures vary at Meter Readings must be converted to Standard conditions for billing Application of Correction Factors for Pressure and/or Temperature Introduction: Most gas meters measure the volume of gas at existing line conditions of pressure and temperature. This volume is usually referred to as displaced volume or actual volume (VA). The value of the gas (i.e., heat content) is referred to in gas measurement as the standard volume (VS) or volume at standard conditions of pressure and temperature. Since gases are compressible fluids, a meter that is measuring gas at two (2) atmospheres will have twice the capacity that it would have if the gas is being measured at one (1) atmosphere. (Note: One atmosphere is the pressure exerted by the air around us. This value is normally 14.696 psi absolute pressure at sea level, or 29.92 inches of mercury.) This fact is referred to as Boyle’s Law which states, “Under constant tempera- ture conditions, the volume of gas is inversely proportional to the ratio of the change in absolute pressures”. This can be expressed mathematically as: * P1 V1 = P2 V2 or P1 = V2 P2 V1 Charles’ Law states that, “Under constant pressure conditions, the volume of gas is directly proportional to the ratio of the change in absolute temperature”. Or mathematically, * V1 = V2 or V1 T2 = V2 T1 T1 T2 Gas meters are normally rated in terms of displaced cubic feet per hour.