Overexpressed Somatic Alleles Are Enriched in Functional Elements in Breast Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluation of Potential Biomarkers for Ewing´S Sarcoma As Cargo of Tumor Derived Exosomes
TECHNISCHE UNIVERSITÄT MÜNCHEN Klinik und Poliklinik für Kinder- und Jugendmedizin Klinikum rechts der Isar Direktor: Univ.-Prof. Dr. St. Burdach Evaluation of potential biomarkers for Ewing´s sarcoma as cargo of tumor derived exosomes Isabella Viktoria Miller Vollständiger Abdruck der von der Fakultät für Medizin der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Medizin genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. E. J. Rummeny Prüfer der Dissertation: 1. Univ.-Prof. Dr. St. Burdach 2. Priv.-Doz. Dr. J. M. E. Teichert-von Lüttichau 3. Univ.-Prof. Dr. A. Krackhardt Die Dissertation wurde am 31.10.2014 bei der Technischen Universität München eingereicht und durch die Fakultät für Medizin am 03.02.2016 angenommen. Contents List of Abbreviations 6 Summary 8 1 Introduction 9 1.1 Ewing´s sarcoma . .9 1.1.1 The Ewing family of tumors . .9 1.1.2 RNA based markers for subclinical disease and their limitations . 10 1.2 Exosomes . 11 1.2.1 Biogenesis and classification . 11 1.2.2 Communication pathways . 12 1.2.3 Roles in tumorigenesis . 14 1.2.3.1 Immunosuppression . 15 1.2.3.2 Angiogenesis . 15 1.2.3.3 Modulation of the microenvironment . 15 1.2.3.4 Metastasis . 16 1.2.3.5 Drug resistance . 17 1.2.4 Diagnostic implications as tumor markers . 17 1.3 Research objectives . 24 1.3.1 Aim of the project . 24 1.3.2 Key questions . 24 2 Materials and Methods 25 2.1 Materials . 25 2.1.1 List of Manufacturers . 25 2.1.2 General materials . -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

ATA33632-NBPF3 Rabbit Polyclonal Antibody
ATAGENIX LABORATORIES Catalog Number:ATA33632 NBPF3 rabbit Polyclonal Antibody Product overview product name NBPF3 rabbit Polyclonal Antibody catalog No. ATA33632 Category Primary antibodies Host Rabbit Species specificity Human Tested applications WB,ELISA Clonality Polyclonal Conjugation Unconjugated Immunogen Synthesized peptide derived from human protein . at AA range: 410-490 Product performance Form Liquid Storage Use a manual defrost freezer and avoid repeated freeze thaw cycles. Store at 4 °C for frequent use. Store at -20 to -80 °C for twelve months from the date of receipt. Purity The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen. Dilution range WB 1:500-2000 ELISA 1:5000-20000 Product background neuroblastoma breakpoint family member 3(NBPF3) Homo sapiens This gene is a member of the neuroblastoma breakpoint family (NBPF) which consists of dozens of recently duplicated genes primarily located in segmental duplications on human chromosome 1. This gene family has experienced its greatest expansion within the human lineage and has expanded, to a lesser extent, among primates in general. Members of this gene family are characterized by tandemly repeated copies of DUF1220 protein domains. DUF1220 copy number variations in human chromosomal region 1q21.1, where most DUF1220 domains are located, have been implicated in a number of developmental and neurogenetic diseases such as microcephaly, macrocephaly, autism, schizophrenia, mental retardation, congenital heart disease, neuroblastoma, and congenital kidney and urinary tract anomalies. Altered expression of some gene family members is associated with several types of cancer. This gene fami Web:www.atagenix.com E-mail: [email protected] Tel: 027-87433958. -

Evidence for Somatic Gene Conversion and Deletion in Bipo- Lar Disorder, Crohn's Disease, Coronary Artery Disease, Hypertensio
Evidence for Somatic Gene Conversion and Deletion in Bipo- lar Disorder, Crohn’s Disease, Coronary Artery Disease, Hypertension, Rheumatoid Arthritis, Type-1 Diabetes, and Type-2 Diabetes Kenneth Andrew Ross∗ Department of Computer Science, Columbia University, New York NY 10027, USA Email: [email protected]; ∗Corresponding author Additional File 1 No-Calls in Chiamo When the converting sequence possesses a B allele, the spread from the AA cluster to the AB cluster will be twice that of the spread from the AB cluster to the BB cluster, as explained in the main text. When A is the more common allele, points between AA and AB will be the most common between-cluster points and will frequently be no-calls. As a result, the B allele frequency will appear to increase among called genotypes. When A is the rarer allele, the higher spread from AA to AB may be counterbalanced by a higher number of AB genotypes relative to AA genotypes. In this case, Chiamo may display a bias in either direction, identifying a smaller number of more distant points as no-calls, or a larger number of less-distant points as no-calls. In some cases, both groups of points may be identified as no-calls, with only a minor resulting bias. Occasionally, two clusters will appear so close together in a population that Chiamo redefines the clusters for that population. In that case, a subset of points from one cluster will be called as part of the neighboring cluster, leading to very large changes in apparent genotype frequencies. -

Genome-Wide Association Study on Serum Alkaline Phosphatase Levels in a Chinese Population
Genome-wide association study on serum alkaline phosphatase levels in a Chinese population The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Li, J., L. Gui, C. Wu, Y. He, L. Zhou, H. Guo, J. Yuan, et al. 2013. “Genome-wide association study on serum alkaline phosphatase levels in a Chinese population.” BMC Genomics 14 (1): 684. doi:10.1186/1471-2164-14-684. http:// dx.doi.org/10.1186/1471-2164-14-684. Published Version doi:10.1186/1471-2164-14-684 Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:11879220 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Li et al. BMC Genomics 2013, 14:684 http://www.biomedcentral.com/1471-2164/14/684 RESEARCH ARTICLE Open Access Genome-wide association study on serum alkaline phosphatase levels in a Chinese population Jun Li1†, Lixuan Gui1†, Chen Wu2, Yunfeng He1, Li Zhou1,5, Huan Guo1, Jing Yuan1, Binyao Yang1, Xiayun Dai1, Qifei Deng1, Suli Huang1, Lei Guan1, Die Hu1, Siyun Deng1, Tian Wang1, Jiang Zhu3, Xinwen Min3, Mingjian Lang3, Dongfeng Li3, Handong Yang3, Frank B Hu4, Dongxin Lin2, Tangchun Wu1 and Meian He1* Abstract Background: Serum alkaline phosphatase (ALP) is a complex phenotype influenced by both genetic and environmental factors. Recent Genome-Wide Association Studies (GWAS) have identified several loci affecting ALP levels; however, such studies in Chinese populations are limited. -

Interlocus Gene Conversion Events Introduce Deleterious Mutations Into at Least 1% of Human Genes Associated with Inherited Disease
Supplemental Material for: Interlocus gene conversion events introduce deleterious mutations into at least 1% of human genes associated with inherited disease Claudio Casola1, Ugne Zekonyte1, Andrew D. Phillips2, David N. Cooper2, and Matthew W. Hahn1,3 1Department of Biology and 3School of Informatics and Computing, Indiana University, Bloomington, IN 47405 2 Institute of Medical Genetics, School of Medicine, Cardiff University, Heath Park, Cardiff CF14 4XN, United Kingdom. Corresponding author: Claudio Casola Department of Biology, Indiana University 1001 East 3rd Street, Bloomington, IN 47405 Phone: 812-856-7016 Fax: 812-855-6705 Email: [email protected] 1 Methods Validation of disease alleles originating from interlocus gene conversion All candidate IGC-derived disease alleles identified by our BLAST searches were further examined to identify high-confidence IGC events. Such alleles required at least one disease-associated mutation plus another mutation to be shared by the IGC donor sequence. In addition, we excluded from our data set those cases where hypermutable CpG sites constituted all the diagnostic mutations. After careful examination of the available literature, we also disregarded several putative IGC events that were deemed likely to represent experimental artifacts. In particular, we identified several cases where primer pairs, which were originally considered to have amplified a single genomic locus or cDNA transcript, also appeared to be capable of amplifying sequences paralogous to the target genes. Such instances included disease alleles from the genes CFC1 (Bamford et al. 2000), GPD2 (Novials et al. 1997) and HLA-DRB1 (Velickovic et al. 2004). In the first two of these cases, we noted that the primer pairs that had been originally designed to amplify the genes specifically, also perfectly matched the paralogous gene CFC1B and an unlinked processed pseudogene of GPD2, respectively. -

The Driver of Extreme Human-Specific Olduvai Repeat Expansion Remains Highly Active in the Human
Genetics: Early Online, published on November 21, 2019 as 10.1534/genetics.119.302782 1 The Driver of Extreme Human-Specific Olduvai Repeat Expansion Remains Highly Active in the Human 2 Genome 3 Ilea E. Heft,1,* Yulia Mostovoy,2,* Michal Levy-Sakin,2 Walfred Ma,2 Aaron J. Stevens,3 Steven 4 Pastor,4 Jennifer McCaffrey,4 Dario Boffelli,5 David I. Martin,5 Ming Xiao,4 Martin A. Kennedy,3 5 Pui-Yan Kwok,2,6,7 and James M. Sikela1 6 7 1Department of Biochemistry and Molecular Genetics, and Human Medical Genetics and 8 Genomics Program, University of Colorado School of Medicine, Aurora CO 80045 9 2Cardiovascular Research Institute, 6Department of Dermatology, and 7Institute for Human 10 Genetics, University of California, San Francisco, San Francisco, CA, USA 11 3Department of Pathology, University of Otago, Christchurch, New Zealand 8140 12 4School of Biomedical Engineering, Drexel University, Philadelphia, PA 19104 13 5Children's Hospital Oakland Research Institute, Oakland, CA, 94609 14 15 Corresponding author: James M. Sikela: [email protected] 16 *Ilea E. Heft and Yulia Mostovoy contributed equally to this article. 17 1 Copyright 2019. 18 Abstract 19 Sequences encoding Olduvai protein domains (formerly DUF1220) show the greatest 20 human lineage-specific increase in copy number of any coding region in the genome and have 21 been associated, in a dosage-dependent manner, with brain size, cognitive aptitude, autism, 22 and schizophrenia. Tandem intragenic duplications of a three-domain block, termed the Olduvai 23 triplet, in four NBPF genes in the chromosomal 1q21.1-.2 region are primarily responsible for 24 the striking human-specific copy number increase. -

High Resolution Measurement of DUF1220 Domain Copy Number from Whole Genome Sequence Data David P
Astling et al. BMC Genomics (2017) 18:614 DOI 10.1186/s12864-017-3976-z METHODOLOGYARTICLE Open Access High resolution measurement of DUF1220 domain copy number from whole genome sequence data David P. Astling1, Ilea E. Heft1, Kenneth L. Jones2 and James M. Sikela1* Abstract Background: DUF1220 protein domains found primarily in Neuroblastoma BreakPoint Family (NBPF) genes show the greatest human lineage-specific increase in copy number of any coding region in the genome. There are 302 haploid copies of DUF1220 in hg38 (~160 of which are human-specific) and the majority of these can be divided into 6 different subtypes (referred to as clades). Copy number changes of specific DUF1220 clades have been associated in a dose-dependent manner with brain size variation (both evolutionarily and within the human population), cognitive aptitude, autism severity, and schizophrenia severity. However, no published methods can directly measure copies of DUF1220 with high accuracy and no method can distinguish between domains within a clade. Results: Here we describe a novel method for measuring copies of DUF1220 domains and the NBPF genes in which they are found from whole genome sequence data. We have characterized the effect that various sequencing and alignment parameters and strategies have on the accuracy and precision of the method and defined the parameters that lead to optimal DUF1220 copy number measurement and resolution. We show that copy number estimates obtained using our read depth approach are highly correlated with those generated by ddPCR for three representative DUF1220 clades. By simulation, we demonstrate that our method provides sufficient resolution to analyze DUF1220 copy number variation at three levels: (1) DUF1220 clade copy number within individual genes and groups of genes (gene-specific clade groups) (2) genome wide DUF1220 clade copies and (3) gene copy number for DUF1220-encoding genes. -

Gene Section Mini Review
Atlas of Genetics and Cytogenetics in Oncology and Haematology OPEN ACCESS JOURNAL AT INIST-CNRS Gene Section Mini Review RAP1GAP (RAP1 GTPase activating protein) Zixing Chen, Xuejun Shao Jiangsu Institute of Hematology, 1st Affiliated Hospital, Soochow University, Suzhou 215006 JS, China (ZC, XS) Published in Atlas Database: January 2010 Online updated version : http://AtlasGeneticsOncology.org/Genes/RAP1GAPID42043ch1p36.html DOI: 10.4267/2042/44888 This work is licensed under a Creative Commons Attribution-Noncommercial-No Derivative Works 2.0 France Licence. © 2010 Atlas of Genetics and Cytogenetics in Oncology and Haematology Identity Function GTPase activator for the nuclear Ras-related regulatory Other names: RAPGAP; RAP1GA1; KIAA0474; protein Rap1, converting it to the putatively inactive RAP1GAP1; RAP1GAPII GDP-bound state (according to Swiss-Prot); Regulation HGNC (Hugo): RAP1GAP of small GTPase-mediated signal transduction. Location: 1p36.12 Homology Local order: From centromere to telomere: NBPF3, The RAP1GAP gene is conserved in cow, mouse, rat, ALPL, RAP1GAP, USP48, HSPG2. zebrafish, fruit fly, mosquito, and C. elegans. DNA/RNA Implicated in Description Solid tumors 25 exons encompassing about 73 kb of genomic DNA. Disease Transcription Papillary thyroid cancer, pancreatic cancer, prostate About 3.334 kb mRNA, and has three transcript cancer, melanoma tumors variant, RAP1 GTPase activating protein isoform a, b, Oncogenesis c. Rap1GAP, which acts as a GTPase activator for the nuclear Ras-related regulatory protein Rap1, was a Protein specific negative regulator of Rap1, and the monomeric G protein Rap1 has been implicated in cancer Description tumorigenesis. It signals to pathways involved in cell 663 amino acids; homodimer and heterodimer with adhesion, migration, and survival. -
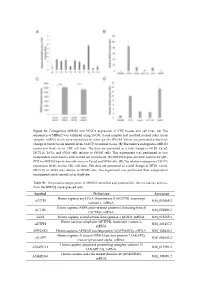
Figure S1. Endogenous MIR45
Figure S1. Endogenous MIR452 and VEGFA expression in CRC tissues and cell lines. (A) The expression of MIR452 was validated using 10 CRC tissue samples and matched normal colon tissue samples. miRNA levels were normalized to colon-specific RNU48. Values are presented as the fold- change in tumor tissue relative levels (ΔΔCT) to normal tissue. (B) The relative endogenous MIR452 expression levels in six CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, LoVo, and SW48 cells relative to SW480 cells. This experiment was performed as two independent experiments, each carried out in triplicate. (C) MIR452 expression level analysis by qRT- PCR for MIR452 transfection efficiency in Caco2 and SW48 cells. (D) The relative endogenous VEGFA expression levels in five CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, or SW48 cells relative to SW480 cells. This experiment was performed three independent experiments, each carried out in duplicate. Table S1. The putative target genes of MIR452 identified and predicted by the microarray analysis from the MIR452 overexpressed cells. Symbol Definition Accession Homo sapiens acyl-CoA thioesterase 8 (ACOT8), transcript ACOT8 NM_005469.2 variant 1, mRNA. Homo sapiens ARP6 actin-related protein 6 homolog (yeast) ACTR6 NM_022496.3 (ACTR6), mRNA. ADI1 Homo sapiens acireductone dioxygenase 1 (ADI1), mRNA. NM_018269.1 Homo sapiens aftiphilin (AFTPH), transcript variant 1, AFTPH NM_203437.2 mRNA. AHNAK2 Homo sapiens AHNAK nucleoprotein 2 (AHNAK2), mRNA. NM_138420.2 Homo sapiens A kinase (PRKA) anchor protein 7 (AKAP7), AKAP7 NM_004842.2 transcript variant alpha, mRNA. Homo sapiens anaphase promoting complex subunit 13 ANAPC13 NM_015391.2 (ANAPC13), mRNA. -

The Hormone-Bound Vitamin D Receptor Regulates Turnover of Target
The hormone-bound vitamin D receptor regulates turnover of target proteins of the SCFFBW7 E3 ligase By Reyhaneh Salehi Tabar Department of Experimental Medicine McGill University Montreal, QC, Canada April 2016 A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of Doctor of Philosophy © Reyhaneh Salehi Tabar 1 Table of Contents Abbreviations ................................................................................................................................................ 7 Abstract ....................................................................................................................................................... 10 Rèsumè ....................................................................................................................................................... 13 Acknowledgements ..................................................................................................................................... 16 Preface ........................................................................................................................................................ 17 Contribution of authors .............................................................................................................................. 18 Chapter 1-Literature review........................................................................................................................ 20 1.1. General introduction and overview of thesis ............................................................................ -

Comprehensive Wellness Report
COMPREHENSIVE WELLNESS REPORT CAsdm1411 UHtest Date of Birth myDNA ID: Pathology No: Reported: 15 May 1990 24505 21628K1Z1R2 14 November 2018 Page | i Things to consider We advise that you consult with your healthcare practitioner before you start any diet program recommended in this report. These recommendations may differ based on your age, height, weight or activity levels. Further, the diet recommendations in this report apply to over 18 years old. Please consult your healthcare practitioner about any diet and lifestyle changes if you are under 18 years of age. The information in this report is not intended to treat, diagnose or cure any medical condition or disease. Page | ii ABOUT THIS REPORT What are genetics and nutrigenomics? How was your report created? Your DNA contains information that determines the Your DNA was extracted from the cheek sample you provided characteristics that are with you at birth. These include hair and and was analyzed at myDNA’s NATA accredited laboratory in eye color, and other characteristics such as how you process Melbourne, Australia. nutrients. This genetic information affects you from the inside. Based on the available information found in the published Your environment, nutritional intake, and lifestyle also play an literature, each gene has been assigned a category according to important role. These factors affect you from the outside. the likely clinical significance. The interaction between nutrients and genes is referred to as The three categories are: nutrigenomics. Least favorable Less favorable Normal/favorable Recent research has revealed the value of nutrigenomics testing for personal use. Individuals who are guided by their genetic It is important to note that this categorization does not relate to profile are more likely to make sustainable, long-term and the exact number of risk variants.