Schönhuth Et Al., 2015)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Inversión Programada 2009
Desarrollo Rural Inversión Programada 2009 Inversión (Pesos) Vertiente Programas / Proyectos % Total Federal Estatal Otras Inversión Normal Estatal Obra Directa y Proyectos Especiales. 32.9 412,113,069 0 361,766,064 50,347,005 SDR - SAGARPA, SDR - SEMARNAT, Concertados Reconversión Productiva y Apoyo a 67.1 839,356,203 446,345,210 178,481,899 214,529,094 MPYMES. Total 100.0 1,251,469,272 446,345,210 540,247,963 264,876,099 Porcentaje de Estructura Financiera 100.0 35.7 43.2 21.2 Fuente: Secretaría de Desarrollo Rural. ADMINISTRACIÓN 2004-2010 139 Desarrollo Rural Inversión Nominal al Campo con Participación de Gobierno del Estado Millones de Año Pesos 1995 215.40 1996 327.70 1997 336.80 1998 448.20 1999 595.70 2000 561.50 2001 662.90 2002 728.60 2003 565.07 2004 878.32 2005 1,028.68 2006 1,422.10 2007 1,203.90 2008 1,302.20 2009 1,251.50 1,422.10 1,302.20 1,203.90 1,251.50 1,028.68 878.32 728.60 662.90 595.70 561.50 448.20 565.07 327.70 336.80 Millones de Pesos 215.40 1995 1996 1997 1998 1999 2000 2001 2002 2003 2004 2005 2006 2007 2008 2009 Fuente: Secretaría de Desarrollo Rural. 140 QUINTO INFORME Desarrollo Rural Programa Dotación Insumos (Semilla y Autoconsumo) octubre 2008 - septiembre 2009 Tipo de Obra Tipo de Obra Municipio SemillaUnidad de Beneficiados Municipio Semilla Unidad de Beneficiados Cantidad Cantidad Medida Medida FRIJOL Toneladas 0.25 1 AVENA Toneladas 0.6 2 AHUMADA MAIZ Toneladas 0.2 1 FRIJOL Toneladas 1.2 9 CASAS GRANDES AVENA Toneladas 5.5 14 MAIZ Toneladas 0.22 3 FRIJOL Toneladas 0.6 9 SORGO Toneladas 1.06 8 ALDAMA -

And Wildlife, 1928-72
Bibliography of Research Publications of the U.S. Bureau of Sport Fisheries and Wildlife, 1928-72 UNITED STATES DEPARTMENT OF THE INTERIOR BUREAU OF SPORT FISHERIES AND WILDLIFE RESOURCE PUBLICATION 120 BIBLIOGRAPHY OF RESEARCH PUBLICATIONS OF THE U.S. BUREAU OF SPORT FISHERIES AND WILDLIFE, 1928-72 Edited by Paul H. Eschmeyer, Division of Fishery Research Van T. Harris, Division of Wildlife Research Resource Publication 120 Published by the Bureau of Sport Fisheries and Wildlife Washington, B.C. 1974 Library of Congress Cataloging in Publication Data Eschmeyer, Paul Henry, 1916 Bibliography of research publications of the U.S. Bureau of Sport Fisheries and Wildlife, 1928-72. (Bureau of Sport Fisheries and Wildlife. Kesource publication 120) Supt. of Docs. no.: 1.49.66:120 1. Fishes Bibliography. 2. Game and game-birds Bibliography. 3. Fish-culture Bibliography. 4. Fishery management Bibliogra phy. 5. Wildlife management Bibliography. I. Harris, Van Thomas, 1915- joint author. II. United States. Bureau of Sport Fisheries and Wildlife. III. Title. IV. Series: United States Bureau of Sport Fisheries and Wildlife. Resource publication 120. S914.A3 no. 120 [Z7996.F5] 639'.9'08s [016.639*9] 74-8411 For sale by the Superintendent of Documents, U.S. Government Printing OfTie Washington, D.C. Price $2.30 Stock Number 2410-00366 BIBLIOGRAPHY OF RESEARCH PUBLICATIONS OF THE U.S. BUREAU OF SPORT FISHERIES AND WILDLIFE, 1928-72 INTRODUCTION This bibliography comprises publications in fishery and wildlife research au thored or coauthored by research scientists of the Bureau of Sport Fisheries and Wildlife and certain predecessor agencies. Separate lists, arranged alphabetically by author, are given for each of 17 fishery research and 6 wildlife research labora tories, stations, investigations, or centers. -

Early Development of the Devils River Minnow, Dionda Diaboli (Cyprinidae)
THE SOUTHWESTERN NATURALIST 52(3):378–385 SEPTEMBER 2007 EARLY DEVELOPMENT OF THE DEVILS RIVER MINNOW, DIONDA DIABOLI (CYPRINIDAE) JULIE HULBERT,TIMOTHY H. BONNER,* JOE N. FRIES,GARY P. GARRETT, AND DAVID R. PENDERGRASS Department of Biology, Texas State University, 601 University Drive, San Marcos, TX 78666 (JH, THB, DRP) National Fish Hatchery and Technology Center, 500 McCarty Lane, San Marcos, TX 78666 (JNF) Texas Parks and Wildlife Department, 5103 Junction Highway, Ingram, TX 78025 (GPG) *Correspondent: [email protected] ABSTRACT—The Devils River minnow (Dionda diaboli) coexists with at least 2 congeners and several other cyprinids throughout its range in southern Texas and northern Mexico. Larval and juvenile descriptions are needed to monitor D. diaboli larvae and juveniles as part of recovery efforts for this species of conservation concern. The purpose of this study was to describe and quantify characteristics of early life stages of D. diaboli from hatching to 128 d post hatch to facilitate larval and juvenile identification. Descriptive characters include mid-lateral band of melanophores by Day 8 (.5.1 mm SL; .5.4 mm TL), mid-lateral band of melanophores separate from a rounded caudal spot and lateral snout-to-eye melanophores by Day 16 (.5.8 mm SL; .6.3 mm TL), initial coiling of intestine by Day 32 (.6.2 mm SL; .7.2 mm TL), wedge-shaped caudal spot by Day 64 (.8.7 mm SL; .10.0 mm TL), and melanophores around scale margins and mid-lateral double dashes along lateral line by Day 128 (.13.5 mm SL; .16.0 mm TL). -

Dionda Diaboli), 6 with Implications for Its Conservation…………………………………
ECOLOGICAL PROFILES FOR SELECTED STREAM-DWELLING TEXAS FRESHWATER FISHES IV A Final Report To The Texas Water Development Board Prepared by: Robert J. Edwards Department of Biology University of Texas-Pan American Edinburg, TX 78541 TWDB Contract Number: 95-483-107 30 August 2003 TABLE OF CONTENTS Table of Contents ………………………………………….…………………….. 2 Introduction 3 Scientific Publications Resulting From This TWDB Contract………...………. 3 Discovery of a New Population of Devils River Minnow (Dionda diaboli), 6 With Implications for its Conservation…………………………………. Acknowledgements………………………………………………………..………. 14 Literature Cited……………….………………………………………….……….. 14 2 Introduction A major goal of the Water Development Board's mission are its research, monitoring, and assessment programs designed to minimize the effects of water development projects on the affected native aquatic fauna and to maintain the quality and availability of instream habitats for the use of dependent aquatic resources. The instream flows necessary for the successful survival, growth and reproduction of affected aquatic life are a major concern. Unfortunately, instream flow data with respect to the ecological requirements of Texas riverine fishes are largely unknown. While some information can be found in the published literature, a substantial but unknown quantity of information is also present in various agencies and research museums around the state. In order to minimize the disruptions to the native fauna, quantitative and qualitative information concerning life histories, survival, -

Information on the NCWRC's Scientific Council of Fishes Rare
A Summary of the 2010 Reevaluation of Status Listings for Jeopardized Freshwater Fishes in North Carolina Submitted by Bryn H. Tracy North Carolina Division of Water Resources North Carolina Department of Environment and Natural Resources Raleigh, NC On behalf of the NCWRC’s Scientific Council of Fishes November 01, 2014 Bigeye Jumprock, Scartomyzon (Moxostoma) ariommum, State Threatened Photograph by Noel Burkhead and Robert Jenkins, courtesy of the Virginia Division of Game and Inland Fisheries and the Southeastern Fishes Council (http://www.sefishescouncil.org/). Table of Contents Page Introduction......................................................................................................................................... 3 2010 Reevaluation of Status Listings for Jeopardized Freshwater Fishes In North Carolina ........... 4 Summaries from the 2010 Reevaluation of Status Listings for Jeopardized Freshwater Fishes in North Carolina .......................................................................................................................... 12 Recent Activities of NCWRC’s Scientific Council of Fishes .................................................. 13 North Carolina’s Imperiled Fish Fauna, Part I, Ohio Lamprey .............................................. 14 North Carolina’s Imperiled Fish Fauna, Part II, “Atlantic” Highfin Carpsucker ...................... 17 North Carolina’s Imperiled Fish Fauna, Part III, Tennessee Darter ...................................... 20 North Carolina’s Imperiled Fish Fauna, Part -

Endangered Species
FEATURE: ENDANGERED SPECIES Conservation Status of Imperiled North American Freshwater and Diadromous Fishes ABSTRACT: This is the third compilation of imperiled (i.e., endangered, threatened, vulnerable) plus extinct freshwater and diadromous fishes of North America prepared by the American Fisheries Society’s Endangered Species Committee. Since the last revision in 1989, imperilment of inland fishes has increased substantially. This list includes 700 extant taxa representing 133 genera and 36 families, a 92% increase over the 364 listed in 1989. The increase reflects the addition of distinct populations, previously non-imperiled fishes, and recently described or discovered taxa. Approximately 39% of described fish species of the continent are imperiled. There are 230 vulnerable, 190 threatened, and 280 endangered extant taxa, and 61 taxa presumed extinct or extirpated from nature. Of those that were imperiled in 1989, most (89%) are the same or worse in conservation status; only 6% have improved in status, and 5% were delisted for various reasons. Habitat degradation and nonindigenous species are the main threats to at-risk fishes, many of which are restricted to small ranges. Documenting the diversity and status of rare fishes is a critical step in identifying and implementing appropriate actions necessary for their protection and management. Howard L. Jelks, Frank McCormick, Stephen J. Walsh, Joseph S. Nelson, Noel M. Burkhead, Steven P. Platania, Salvador Contreras-Balderas, Brady A. Porter, Edmundo Díaz-Pardo, Claude B. Renaud, Dean A. Hendrickson, Juan Jacobo Schmitter-Soto, John Lyons, Eric B. Taylor, and Nicholas E. Mandrak, Melvin L. Warren, Jr. Jelks, Walsh, and Burkhead are research McCormick is a biologist with the biologists with the U.S. -

ECOLOGY of NORTH AMERICAN FRESHWATER FISHES
ECOLOGY of NORTH AMERICAN FRESHWATER FISHES Tables STEPHEN T. ROSS University of California Press Berkeley Los Angeles London © 2013 by The Regents of the University of California ISBN 978-0-520-24945-5 uucp-ross-book-color.indbcp-ross-book-color.indb 1 44/5/13/5/13 88:34:34 AAMM uucp-ross-book-color.indbcp-ross-book-color.indb 2 44/5/13/5/13 88:34:34 AAMM TABLE 1.1 Families Composing 95% of North American Freshwater Fish Species Ranked by the Number of Native Species Number Cumulative Family of species percent Cyprinidae 297 28 Percidae 186 45 Catostomidae 71 51 Poeciliidae 69 58 Ictaluridae 46 62 Goodeidae 45 66 Atherinopsidae 39 70 Salmonidae 38 74 Cyprinodontidae 35 77 Fundulidae 34 80 Centrarchidae 31 83 Cottidae 30 86 Petromyzontidae 21 88 Cichlidae 16 89 Clupeidae 10 90 Eleotridae 10 91 Acipenseridae 8 92 Osmeridae 6 92 Elassomatidae 6 93 Gobiidae 6 93 Amblyopsidae 6 94 Pimelodidae 6 94 Gasterosteidae 5 95 source: Compiled primarily from Mayden (1992), Nelson et al. (2004), and Miller and Norris (2005). uucp-ross-book-color.indbcp-ross-book-color.indb 3 44/5/13/5/13 88:34:34 AAMM TABLE 3.1 Biogeographic Relationships of Species from a Sample of Fishes from the Ouachita River, Arkansas, at the Confl uence with the Little Missouri River (Ross, pers. observ.) Origin/ Pre- Pleistocene Taxa distribution Source Highland Stoneroller, Campostoma spadiceum 2 Mayden 1987a; Blum et al. 2008; Cashner et al. 2010 Blacktail Shiner, Cyprinella venusta 3 Mayden 1987a Steelcolor Shiner, Cyprinella whipplei 1 Mayden 1987a Redfi n Shiner, Lythrurus umbratilis 4 Mayden 1987a Bigeye Shiner, Notropis boops 1 Wiley and Mayden 1985; Mayden 1987a Bullhead Minnow, Pimephales vigilax 4 Mayden 1987a Mountain Madtom, Noturus eleutherus 2a Mayden 1985, 1987a Creole Darter, Etheostoma collettei 2a Mayden 1985 Orangebelly Darter, Etheostoma radiosum 2a Page 1983; Mayden 1985, 1987a Speckled Darter, Etheostoma stigmaeum 3 Page 1983; Simon 1997 Redspot Darter, Etheostoma artesiae 3 Mayden 1985; Piller et al. -

Geological Survey of Alabama Calibration of The
GEOLOGICAL SURVEY OF ALABAMA Berry H. (Nick) Tew, Jr. State Geologist WATER INVESTIGATIONS PROGRAM CALIBRATION OF THE INDEX OF BIOTIC INTEGRITY FOR THE SOUTHERN PLAINS ICHTHYOREGION IN ALABAMA OPEN-FILE REPORT 0908 by Patrick E. O'Neil and Thomas E. Shepard Prepared in cooperation with the Alabama Department of Environmental Management and the Alabama Department of Conservation and Natural Resources Tuscaloosa, Alabama 2009 TABLE OF CONTENTS Abstract ............................................................ 1 Introduction.......................................................... 1 Acknowledgments .................................................... 6 Objectives........................................................... 7 Study area .......................................................... 7 Southern Plains ichthyoregion ...................................... 7 Methods ............................................................ 8 IBI sample collection ............................................. 8 Habitat measures............................................... 10 Habitat metrics ........................................... 12 The human disturbance gradient ................................... 15 IBI metrics and scoring criteria..................................... 19 Designation of guilds....................................... 20 Results and discussion................................................ 22 Sampling sites and collection results . 22 Selection and scoring of Southern Plains IBI metrics . 41 1. Number of native species ................................ -

Kansas Stream Fishes
A POCKET GUIDE TO Kansas Stream Fishes ■ ■ ■ ■ ■ ■ ■ ■ ■ ■ By Jessica Mounts Illustrations © Joseph Tomelleri Sponsored by Chickadee Checkoff, Westar Energy Green Team, Kansas Department of Wildlife, Parks and Tourism, Kansas Alliance for Wetlands & Streams, and Kansas Chapter of the American Fisheries Society Published by the Friends of the Great Plains Nature Center Table of Contents • Introduction • 2 • Fish Anatomy • 3 • Species Accounts: Sturgeons (Family Acipenseridae) • 4 ■ Shovelnose Sturgeon • 5 ■ Pallid Sturgeon • 6 Minnows (Family Cyprinidae) • 7 ■ Southern Redbelly Dace • 8 ■ Western Blacknose Dace • 9 ©Ryan Waters ■ Bluntface Shiner • 10 ■ Red Shiner • 10 ■ Spotfin Shiner • 11 ■ Central Stoneroller • 12 ■ Creek Chub • 12 ■ Peppered Chub / Shoal Chub • 13 Plains Minnow ■ Silver Chub • 14 ■ Hornyhead Chub / Redspot Chub • 15 ■ Gravel Chub • 16 ■ Brassy Minnow • 17 ■ Plains Minnow / Western Silvery Minnow • 18 ■ Cardinal Shiner • 19 ■ Common Shiner • 20 ■ Bigmouth Shiner • 21 ■ • 21 Redfin Shiner Cover Photo: Photo by Ryan ■ Carmine Shiner • 22 Waters. KDWPT Stream ■ Golden Shiner • 22 Survey and Assessment ■ Program collected these Topeka Shiner • 23 male Orangespotted Sunfish ■ Bluntnose Minnow • 24 from Buckner Creek in Hodgeman County, Kansas. ■ Bigeye Shiner • 25 The fish were catalogued ■ Emerald Shiner • 26 and returned to the stream ■ Sand Shiner • 26 after the photograph. ■ Bullhead Minnow • 27 ■ Fathead Minnow • 27 ■ Slim Minnow • 28 ■ Suckermouth Minnow • 28 Suckers (Family Catostomidae) • 29 ■ River Carpsucker • -
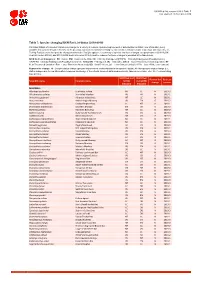
Table 7: Species Changing IUCN Red List Status (2018-2019)
IUCN Red List version 2019-3: Table 7 Last Updated: 10 December 2019 Table 7: Species changing IUCN Red List Status (2018-2019) Published listings of a species' status may change for a variety of reasons (genuine improvement or deterioration in status; new information being available that was not known at the time of the previous assessment; taxonomic changes; corrections to mistakes made in previous assessments, etc. To help Red List users interpret the changes between the Red List updates, a summary of species that have changed category between 2018 (IUCN Red List version 2018-2) and 2019 (IUCN Red List version 2019-3) and the reasons for these changes is provided in the table below. IUCN Red List Categories: EX - Extinct, EW - Extinct in the Wild, CR - Critically Endangered [CR(PE) - Critically Endangered (Possibly Extinct), CR(PEW) - Critically Endangered (Possibly Extinct in the Wild)], EN - Endangered, VU - Vulnerable, LR/cd - Lower Risk/conservation dependent, NT - Near Threatened (includes LR/nt - Lower Risk/near threatened), DD - Data Deficient, LC - Least Concern (includes LR/lc - Lower Risk, least concern). Reasons for change: G - Genuine status change (genuine improvement or deterioration in the species' status); N - Non-genuine status change (i.e., status changes due to new information, improved knowledge of the criteria, incorrect data used previously, taxonomic revision, etc.); E - Previous listing was an Error. IUCN Red List IUCN Red Reason for Red List Scientific name Common name (2018) List (2019) change version Category -

Summary Report of Freshwater Nonindigenous Aquatic Species in U.S
Summary Report of Freshwater Nonindigenous Aquatic Species in U.S. Fish and Wildlife Service Region 4—An Update April 2013 Prepared by: Pam L. Fuller, Amy J. Benson, and Matthew J. Cannister U.S. Geological Survey Southeast Ecological Science Center Gainesville, Florida Prepared for: U.S. Fish and Wildlife Service Southeast Region Atlanta, Georgia Cover Photos: Silver Carp, Hypophthalmichthys molitrix – Auburn University Giant Applesnail, Pomacea maculata – David Knott Straightedge Crayfish, Procambarus hayi – U.S. Forest Service i Table of Contents Table of Contents ...................................................................................................................................... ii List of Figures ............................................................................................................................................ v List of Tables ............................................................................................................................................ vi INTRODUCTION ............................................................................................................................................. 1 Overview of Region 4 Introductions Since 2000 ....................................................................................... 1 Format of Species Accounts ...................................................................................................................... 2 Explanation of Maps ................................................................................................................................ -

Molecular Systematics of Western North American Cyprinids (Cypriniformes: Cyprinidae)
Zootaxa 3586: 281–303 (2012) ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ ZOOTAXA Copyright © 2012 · Magnolia Press Article ISSN 1175-5334 (online edition) urn:lsid:zoobank.org:pub:0EFA9728-D4BB-467E-A0E0-0DA89E7E30AD Molecular systematics of western North American cyprinids (Cypriniformes: Cyprinidae) SUSANA SCHÖNHUTH 1, DENNIS K. SHIOZAWA 2, THOMAS E. DOWLING 3 & RICHARD L. MAYDEN 1 1 Department of Biology, Saint Louis University, 3507 Laclede Avenue, St. Louis, MO 63103, USA. E-mail S.S: [email protected] ; E-mail RLM: [email protected] 2 Department of Biology and Curator of Fishes, Monte L. Bean Life Science Museum, Brigham Young University, Provo, UT 84602, USA. E-mail: [email protected] 3 School of Life Sciences, Arizona State University, Tempe, AZ 85287-4501, USA. E-mail: [email protected] Abstract The phylogenetic or evolutionary relationships of species of Cypriniformes, as well as their classification, is in a era of flux. For the first time ever, the Order, and constituent Families are being examined for relationships within a phylogenetic context. Relevant findings as to sister-group relationships are largely being inferred from analyses of both mitochondrial and nuclear DNA sequences. Like the vast majority of Cypriniformes, due to an overall lack of any phylogenetic investigation of these fishes since Hennig’s transformation of the discipline, changes in hypotheses of relationships and a natural classification of the species should not be of surprise to anyone. Basically, for most taxa no properly supported phylogenetic hypothesis has ever been done; and this includes relationships with reasonable taxon and character sampling of even families and subfamilies.