The Role of Mesenchymal Stromal Cells and Classical Dendritic Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dendritic Cell Subsets in Intestinal Immunity and Inflammation Tian Sun, Albert Nguyen and Jennifer L
Dendritic Cell Subsets in Intestinal Immunity and Inflammation Tian Sun, Albert Nguyen and Jennifer L. Gommerman This information is current as J Immunol 2020; 204:1075-1083; ; of September 28, 2021. doi: 10.4049/jimmunol.1900710 http://www.jimmunol.org/content/204/5/1075 Downloaded from References This article cites 152 articles, 56 of which you can access for free at: http://www.jimmunol.org/content/204/5/1075.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 28, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2020 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Dendritic Cell Subsets in Intestinal Immunity and Inflammation Tian Sun, Albert Nguyen, and Jennifer L. Gommerman The mammalian intestine is a complex environment shaping the ensuing immune response. In this Brief Review,we that is constantly exposed to Ags derived from food, review the different types of classical DC (cDC) located in the microbiota, and metabolites. -

Dendritic Cell Progenitor Trafficking and Identification and Functional Analyses of Dendritic Cells with Distinct Developmental Origin
Dissertation zum Erwerb des Doctor of Philosophy (Ph.D.) an der Medizinischen Fakultät der Ludwig-Maximilians-Universität zu München Dendritic cell progenitor trafficking and identification and functional analyses of dendritic cells with distinct developmental origin vorgelegt von: Johanna Marie Salvermoser aus: Pfaffenhofen an der Ilm Jahr: 2020 _____________________ First supervisor: Prof. Dr. Barbara Schraml Second supervisor: Prof. Dr. Anne Krug Third supervisor: Prof. Dr. Christian Schulz Fourth supervisor: PD. Dr. rer. nat. Caspar Ohnmacht Dean: Prof. Dr. med. dent. Reinhard Hickel Datum der Verteidigung: 09.03.2020 2 Abstract ABSTRACT Conventional dendritic cells (cDCs), are the major antigen-presenting cell type that bridges the innate and adaptive immune system. DCs are constantly replenished from myeloid bone marrow progenitors which latest stage, pre-cDC, leave the BM, seeds the peripheral tissues and further differentiates into two functionally and developmentally distinct subsets, cDC1 and cDC2. This study aimed to investigate DC development by assessing the trafficking of pre-cDCs and by analyzing the effect of a specific depletion of DC progenitors. The signals that regulate the recruitment of pre-cDCs to different peripheral organs are poorly understood. Therefore, this study aimed to identify pre-cDCs in different peripheral organs and to find differences in expression pattern of trafficking receptors. In this study 39 trafficking receptors have been identified to be expressed on pre-cDCs of the analysed tissues and showed differences in the expression patterns between peripheral organs. These receptors are interesting candidates to further study differences in the recruitment of pre-cDCs to different peripheral tissues This can provide possibilities to influence the recruitment of pre-cDCs in certain diseases, where the replenishment of cDCs is accelerated. -

Targeting Dendritic Cells with Antigen-Delivering Antibodies for Amelioration of Autoimmunity in Animal Models of Multiple Sclerosis and Other Autoimmune Diseases
antibodies Review Targeting Dendritic Cells with Antigen-Delivering Antibodies for Amelioration of Autoimmunity in Animal Models of Multiple Sclerosis and Other Autoimmune Diseases Courtney A. Iberg and Daniel Hawiger * Department of Molecular Microbiology and Immunology, Saint Louis University School of Medicine, Doisy Research Center, 1205 Carr Lane, St. Louis, MO 63104, USA; [email protected] * Correspondence: [email protected] Received: 31 March 2020; Accepted: 30 April 2020; Published: 15 June 2020 Abstract: The specific targeting of dendritic cells (DCs) using antigen-delivering antibodies has been established to be a highly efficient protocol for the induction of tolerance and protection from autoimmune processes in experimental autoimmune encephalomyelitis (EAE), a model of multiple sclerosis (MS), as well as in some other animal disease models. As the specific mechanisms of such induced tolerance are being investigated, the newly gained insights may also possibly help to design effective treatments for patients. Here we review approaches applied for the amelioration of autoimmunity in animal models based on antibody-mediated targeting of self-antigens to DCs. Further, we discuss relevant mechanisms of immunological tolerance that underlie such approaches, and we also offer some future perspectives for the application of similar methods in certain related disease settings such as transplantation. Keywords: dendritic cells; tolerance; antigen targeting; chimeric antibodies; autoimmunity; multiple sclerosis; diabetes 1. Introduction Over one hundred years ago, Paul Ehrlich coined the term “horror autotoxicus” to define an immune attack against an organism’s healthy tissues [1]. Since then, our knowledge of the complex mechanisms of the immune system as well as our understanding of the pathogenesis of specific autoimmune diseases have grown tremendously. -

Single-Cell RNA Sequencing Demonstrates the Molecular and Cellular Reprogramming of Metastatic Lung Adenocarcinoma
ARTICLE https://doi.org/10.1038/s41467-020-16164-1 OPEN Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma Nayoung Kim 1,2,3,13, Hong Kwan Kim4,13, Kyungjong Lee 5,13, Yourae Hong 1,6, Jong Ho Cho4, Jung Won Choi7, Jung-Il Lee7, Yeon-Lim Suh8,BoMiKu9, Hye Hyeon Eum 1,2,3, Soyean Choi 1, Yoon-La Choi6,10,11, Je-Gun Joung1, Woong-Yang Park 1,2,6, Hyun Ae Jung12, Jong-Mu Sun12, Se-Hoon Lee12, ✉ ✉ Jin Seok Ahn12, Keunchil Park12, Myung-Ju Ahn 12 & Hae-Ock Lee 1,2,3,6 1234567890():,; Advanced metastatic cancer poses utmost clinical challenges and may present molecular and cellular features distinct from an early-stage cancer. Herein, we present single-cell tran- scriptome profiling of metastatic lung adenocarcinoma, the most prevalent histological lung cancer type diagnosed at stage IV in over 40% of all cases. From 208,506 cells populating the normal tissues or early to metastatic stage cancer in 44 patients, we identify a cancer cell subtype deviating from the normal differentiation trajectory and dominating the metastatic stage. In all stages, the stromal and immune cell dynamics reveal ontological and functional changes that create a pro-tumoral and immunosuppressive microenvironment. Normal resident myeloid cell populations are gradually replaced with monocyte-derived macrophages and dendritic cells, along with T-cell exhaustion. This extensive single-cell analysis enhances our understanding of molecular and cellular dynamics in metastatic lung cancer and reveals potential diagnostic and therapeutic targets in cancer-microenvironment interactions. 1 Samsung Genome Institute, Samsung Medical Center, Seoul 06351, Korea. -

View Dendritic Cell Development Poster
Overview of Dendritic Cell Development Lineage–, CD45+, Common CD117 (c-kit) Common MHCII+, CD11c+ – + CD207 (Langerin) Myeloid CD117 (c-kit) Lineage , CD45 , Myeloid Progenitor MHCII (HLA-DR)+, CD11c+ Progenitor CD324 (E-Cadherin) Human Mouse CD326 (EpCAM) CD207 (Langerin) TGFb1 Cells CD11b, CD115 Cells CD14 Monocyte CD324 (E-Cadherin) Monocyte M-CSF CD11b – + Ly6C Langerhans CD24 Lineage , CD45 , M-CSF CD326 (EpCAM) MHCII (HLA-DR)+, CD11c+ Langerhans CD11blo Zbtb46– Cells CD172a (Sirp-α) CD16 CD1ahi, CD1c CD205 (DEC-205) Cells CSF F4/80 CD64 CD172a (Sirp-α) Lineage–, CD45+, FLT3L TLR3, TLR11 CD1a, CD1c Inflammatory CD369 (Dectin-1/CLEC7A) MHCII+, CD11c+ +/– CSF IL-15 CD8–, CD14– CD11b, CD14 CD371 (CLEC12A) CD64 Monocyte- FLT3L Inflammatory CD370 (Clec9a)– CD172a (Sirp-α) IL-15 CLEC6A CD11b derived lo Monocyte- CD206, CD209 (DC-SIGN) TLR1, TLR2, TLR3 , TLR6 CD209a (DC-SIGN) CD367 (DCIR/CLEC4A) DCs CD14– CD272 (BTLA)lo derived CD369 (Dectin-1/CLEC7A) DCs Common Ly-6C – + CD371 (CLEC12A) CD117 (c-kit) Lineage , CD45 , IL-1β, IL-6, IL-10, TLR1-6, TLR7-8, TLR10 Dendritic + lo CLEC6A – – CD135/FLT3 MHCII , CD11c IL-12, IL-23, TNF CD8a , CD14 IL-1β, IL-6 IL-10, Precursor TLR3lo, TLR4, TLR7, TLR8 CD45R (B220) IL-12, IL-23, TNF Plasmacytoid CD207 (Langerin)– Cells CD317 (BST-2) Common Lineage–, CD45+, FLT3L DCs Lineage–, CD45+, + Ly6C + lo/– CD207 IFN Type I + + Dendritic CD135/FLT3 MHCII (HLA-DR) , CD11c Lineage–, CD45+, IRF7, IRF8, BATF3hi Siglec-H MHCII (HLA-DR) , CD11c hi – + CD123 + + Dermal SpiB, Zbtb46 CD1a, CD64 CD1a Precursor CD117 (c-kit) -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -
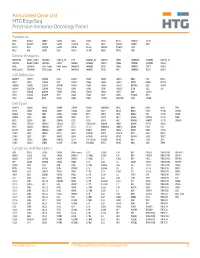
Annotated Gene List HTG Edgeseq Precision Immuno-Oncology Panel
Annotated Gene List HTG EdgeSeq Precision Immuno-Oncology Panel Apoptosis APAF1 BCL2L1 CARD11 CASP4 CD5L FADD KSR2 OPTN SAMD12 TCF19 BAX BCL2L11 CASP1 CASP5 CORO1A FAS LRG1 PLA2G6 SAMD9 XAF1 BCL10 BCL6 CASP10 CASP8 DAPK2 FASLG MECOM PYCARD SPOP BCL2 BID CASP3 CAV1 DAPL1 GLIPR1 MELK RIPK2 TBK1 Cancer Antigens ANKRD30A BAGE2_BAGE3 CEACAM6 CTAG1A_1B LIPE MAGEA3_A6 MAGEC2 PAGE3 SPANXACD SPANXN4 XAGE1B_1E ARMCX6 BAGE4_BAGE5 CEACAM8 CTAG2 MAGEA1 MAGEA4 MTFR2 PAGE4 SPANXB1 SPANXN5 XAGE2 BAGE CEACAM1 CT45_family GAGE_family MAGEA10 MAGEB2 PAGE1 PAGE5 SPANXN1 SYCP1 XAGE3 BAGE_family CEACAM5 CT47_family HPN MAGEA12 MAGEC1 PAGE2 PBK SPANXN3 TEX14 XAGE5 Cell Adhesion ADAM17 CDH15 CLEC5A DSG3 ICAM2 ITGA5 ITGB2 LAMC3 MBL2 PVR UPK2 ADD2 CDH5 CLEC6A DST ICAM3 ITGA6 ITGB3 LAMP1 MTDH RRAS2 UPK3A ADGRE5 CLDN3 CLEC7A EPCAM ICAM4 ITGAE ITGB4 LGALS1 NECTIN2 SELE VCAM1 ALCAM CLEC12A CLEC9A FBLN1 ITGA1 ITGAL ITGB7 LGALS3 OCLN SELL ZYX CD63 CLEC2B DIAPH3 FXYD5 ITGA2 ITGAM ITLN2 LYVE1 OLR1 SELPLG CD99 CLEC4A DLGAP5 IBSP ITGA3 ITGAX JAML M6PR PECAM1 THY1 CDH1 CLEC4C DSC3 ICAM1 ITGA4 ITGB1 L1CAM MADCAM1 PKP1 UNC5D Cell Cycle ANAPC1 CCND3 CDCA5 CENPH CNNM1 ESCO2 HORMAD2 KIF2C MELK ORC6 SKA3 TPX2 ASPM CCNE1 CDCA8 CENPI CNTLN ESPL1 IKZF1 KIF4A MND1 PATZ1 SP100 TRIP13 AURKA CCNE2 CDK1 CENPL CNTLN ETS1 IKZF2 KIF5C MYBL2 PIF1 SP110 TROAP AURKB CCNF CDK4 CENPU DBF4 ETS2 IKZF3 KIFC1 NCAPG PIMREG SPC24 TUBB BEX1 CDC20 CDK6 CENPW E2F2 EZH2 IKZF4 KNL1 NCAPG2 PKMYT1 SPC25 ZWILCH BEX2 CDC25A CDKN1A CEP250 E2F7 GADD45GIP1 KDM5B LMNA NCAPH POC1A SPDL1 BUB1 CDC25C CDKN1B CEP55 ECT2 -

Human Clec9a Alexa Fluor® 488-Conjugated Antibody
Human CLEC9a Alexa Fluor® 488-conjugated Antibody Monoclonal Mouse IgG1 Clone # 683409 Catalog Number: FAB6049G 100 Tests DESCRIPTION Species Reactivity Human Specificity Detects human CLEC9a in direct ELISAs. In direct ELISAs, no crossreactivity with recombinant human (rh) CLEC1, 2, 2A, 3B, 10A, 12B, 14A, rhCD302/CLEC13a, rhMICL, or recombinant mouse CLEC9a is observed. Source Monoclonal Mouse IgG1 Clone # 683409 Purification Protein A or G purified from hybridoma culture supernatant Immunogen Mouse myeloma cell line NS0derived recombinant human CLEC9a Lys57Val241 Accession # Q6UXN8 Conjugate Alexa Fluor 488 Excitation Wavelength: 488 nm Emission Wavelength: 515545 nm Formulation Supplied in a saline solution containing BSA and Sodium Azide. See Certificate of Analysis for details. *Contains <0.1% Sodium Azide, which is not hazardous at this concentration according to GHS classifications. Refer to the Safety Data Sheet (SDS) for additional information and handling instructions. APPLICATIONS Please Note: Optimal dilutions should be determined by each laboratory for each application. General Protocols are available in the Technical Information section on our website. Recommended Sample Concentration Flow Cytometry 5 µL/106 cells See Below DATA Flow Cytometry Detection of CLEC9a in Human Peripheral Blood Cells by Flow Cytometry. Human peripheral blood cells gated on CD3CD141+ cells were stained with Mouse AntiHuman HLADR PerCP conjugated Monoclonal Antibody (Catalog # FAB4869C) and either (A) Mouse AntiHuman CLEC9a Alexa Fluor® 488conjugated Monoclonal Antibody (Catalog # FAB6049G) or (B) Mouse IgG1 Alexa Fluor 488 Isotype Control (Catalog # IC002G). View our protocol for Staining Membraneassociated Proteins. PREPARATION AND STORAGE Shipping The product is shipped with polar packs. -

Differential Roles for the Adapters Gads and LAT in Platelet Activation by GPVI and CLEC-2
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by University of Birmingham Research Portal University of Birmingham Differential roles for the adapters Gads and LAT in platelet activation by GPVI and CLEC-2. Hughes, Craig; Auger, Jocelyn; McGlade, J; Eble, JA; Pearce, AC; Watson, Steve DOI: 10.1111/j.1538-7836.2008.03166.x Citation for published version (Harvard): Hughes, C, Auger, J, McGlade, J, Eble, JA, Pearce, AC & Watson, S 2008, 'Differential roles for the adapters Gads and LAT in platelet activation by GPVI and CLEC-2.', Journal of Thrombosis and Haemostasis, vol. 6, no. 12, pp. 2152-9. https://doi.org/10.1111/j.1538-7836.2008.03166.x Link to publication on Research at Birmingham portal General rights Unless a licence is specified above, all rights (including copyright and moral rights) in this document are retained by the authors and/or the copyright holders. The express permission of the copyright holder must be obtained for any use of this material other than for purposes permitted by law. •Users may freely distribute the URL that is used to identify this publication. •Users may download and/or print one copy of the publication from the University of Birmingham research portal for the purpose of private study or non-commercial research. •User may use extracts from the document in line with the concept of ‘fair dealing’ under the Copyright, Designs and Patents Act 1988 (?) •Users may not further distribute the material nor use it for the purposes of commercial gain. Where a licence is displayed above, please note the terms and conditions of the licence govern your use of this document. -

NKT Cell–Driven Enhancement of Antitumor Immunity Induced By
Published OnlineFirst May 3, 2019; DOI: 10.1158/2326-6066.CIR-18-0650 Research Article Cancer Immunology Research NKT Cell–Driven Enhancement of Antitumor Immunity Induced by Clec9a-Targeted Tailorable Nanoemulsion Pui Yeng Lam, Takumi Kobayashi, Megan Soon, Bijun Zeng, Riccardo Dolcetti, Graham Leggatt, Ranjeny Thomas, and Stephen R. Mattarollo Abstract Invariant natural killer T (iNKT) cells are a subset of lym- (Clec9a/OVA/aGC) further enhanced activation of iNKT þ þ phocytes with immune regulatory activity. Their ability to cells, NK cells, CD8a DCs, and polyfunctional CD8 T cells. bridge the innate and adaptive immune systems has been When tested therapeutically against HPVE7-expressing TC-1 studied using the glycolipid ligand a-galactosylceramide tumors, long-term tumor suppression was achieved with a (aGC). To better harness the immune adjuvant properties of single administration of Clec9a/E7 peptide/aGC TNE. Anti- þ iNKT cells to enhance priming of antigen-specific CD8 T cells, tumor activity was correlated with the recruitment of þ we encapsulated both aGC and antigen in a Clec9a-targeted mature DCs, NK cells, and tumor-specific effector CD8 nanoemulsion (TNE) to deliver these molecules to cross- T cells to the tumor-draining lymph node and tumor tissue. þ þ presenting CD8 dendritic cells (DC). We demonstrate that, Thus, Clec9a-TNE codelivery of CD8 T-cell epitopes with even in the absence of exogenous glycolipid, iNKT cells aGC induces alternative helper signals from activated iNKT þ supported the maturation of CD8a DCs to drive efficient cells, elicits innate (iNKT, NK) immunity, and enhances þ þ cross-priming of antigen-specific CD8 T cells upon delivery antitumor CD8 T-cell responses for control of solid of Clec9a/OVA-TNE. -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -

Investigating the Molecular Control of Deer Antler Extract on Articular Cartilage Baojin Yao1, Zhenwei Zhou1, Mei Zhang2, Xiangyang Leng3* and Daqing Zhao1*
Yao et al. Journal of Orthopaedic Surgery and Research (2021) 16:8 https://doi.org/10.1186/s13018-020-02148-w RESEARCH ARTICLE Open Access Investigating the molecular control of deer antler extract on articular cartilage Baojin Yao1, Zhenwei Zhou1, Mei Zhang2, Xiangyang Leng3* and Daqing Zhao1* Abstract Background: Deer antler is considered as a precious traditional Chinese medicinal material and has been widely used to reinforce kidney’s yang, nourish essence, and strengthen bone function. The most prominent bioactive components in deer antler are water-soluble proteins that play potential roles in bone formation and repair. The aim of this study was to explore the molecular control and therapeutic targets of deer antler extract (DAE) on articular cartilage. Methods: DAE was prepared as previously described. All rats were randomly divided into Blank group and DAE group (10 rats per group) after 7-day adaptive feeding. The rats in DAE group were orally administrated with DAE at a dose of 0.2 g/kg per day for 3 weeks, and the rats in Blank group were fed with drinking water. Total RNA was isolated from the articular cartilage of knee joints. RNA sequencing (RNA-seq) experiment combined with quantitative real-time polymerase chain reaction (qRT-PCR) verification assay was carried out to explore the molecular control and therapeutic targets of DAE on articular cartilage. Results: We demonstrated that DAE significantly increased the expression levels of functional genes involved in cartilage formation, growth, and repair and decreased the expression levels of susceptibility genes involved in the pathophysiology of osteoarthritis. Conclusions: DAE might serve as a candidate supplement for maintaining cartilage homeostasis and preventing cartilage degeneration and inflammation.