Differential Expression of the Nuclear-Encoded Mitochondrial Transcriptome in Pediatric Septic Shock
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CD8 T Cell Memory Transition and Maintenance of Functional Foxo1
FoxO1 Controls Effector-to-Memory Transition and Maintenance of Functional CD8 T Cell Memory This information is current as Melba Marie Tejera, Eui Ho Kim, Jeremy A. Sullivan, Erin of October 1, 2021. H. Plisch and M. Suresh J Immunol 2013; 191:187-199; Prepublished online 3 June 2013; doi: 10.4049/jimmunol.1300331 http://www.jimmunol.org/content/191/1/187 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2013/06/03/jimmunol.130033 Material 1.DC1 http://www.jimmunol.org/ References This article cites 52 articles, 7 of which you can access for free at: http://www.jimmunol.org/content/191/1/187.full#ref-list-1 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on October 1, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2013 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology FoxO1 Controls Effector-to-Memory Transition and Maintenance of Functional CD8 T Cell Memory Melba Marie Tejera, Eui Ho Kim, Jeremy A. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

Integrative Genomic Analysis Predicts Causative Cis-Regulatory Mechanisms of the Breast Cancer-Associated Genetic Variant Rs4415084
Author Manuscript Published OnlineFirst on January 19, 2018; DOI: 10.1158/0008-5472.CAN-17-3486 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Cancer Research Mathematical Oncology Title: Integrative genomic analysis predicts causative cis-regulatory mechanisms of the breast cancer-associated genetic variant rs4415084 Authors and affiliations: Yi Zhang1,2*, Mohith Manjunath2*, Shilu Zhang3, Deborah Chasman3, Sushmita Roy3,4, and Jun S. Song2,5 *Equal contribution 1Department of Bioengineering, 2Carl R. Woese Institute for Genomic Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. 3Wisconsin Institute for Discovery, 4Department of Biostatistics and Medical Informatics, University of Wisconsin–Madison, Madison, WI 53792, USA. 5Department of Physics, University of Illinois at Urbana-Champaign, Urbana, IL 61801, USA. Running title: Regulatory genomics of GWAS SNPs Abbreviations: GWAS - genome-wide association study; eQTL - expression quantitative trait loci; ASE - allele-specific expression; TF - transcription factor; LD - linkage disequilibrium; FPKM - fragments per kilobase of transcript per million mapped reads; LCASE - local chromosome allele-specific expression; DHS - DNase I hypersensitive sites; PWM - position weight matrix; TCGA - The Cancer Genome Atlas; ER+ - estrogen receptor positive; TAD - topologically associated domain; MAF - minor allele frequency; RPKM - reads per kilobase of transcript per million mapped reads; SNP - single nucleotide polymorphism; MAPQ - mapping quality; ChIP-seq - chromatin immunoprecipitation sequencing; ASB - allele-specific binding; ncRNA - non-coding RNA; TSS - transcription start site; DNase-seq - DNase I 1 Downloaded from cancerres.aacrjournals.org on September 29, 2021. © 2018 American Association for Cancer Research. Author Manuscript Published OnlineFirst on January 19, 2018; DOI: 10.1158/0008-5472.CAN-17-3486 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -
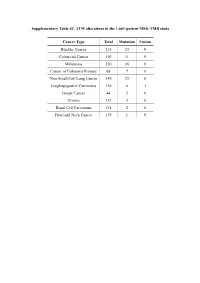
Supplementary Table S1. ATM Alterations in the 1,661-Patient MSK-TMB Study
Supplementary Table S1. ATM alterations in the 1,661-patient MSK-TMB study Cancer Type Total Mutation Fusion Bladder Cancer 215 23 0 Colorectal Cancer 110 11 0 Melanoma 320 26 0 Cancer of Unknown Primary 88 7 0 Non-Small Cell Lung Cancer 350 23 0 Esophagogastric Carcinoma 126 6 1 Breast Cancer 44 2 0 Glioma 117 3 0 Renal Cell Carcinoma 151 2 0 Head and Neck Cancer 139 1 0 Supplementary Table S2. ATM alternations in the 10,945-patient MSK-IMPACT study Cancer Type Total Mutation Deep deletion Amplification Multi-alterations Fusion Small Bowl Cancer 35 7 0 0 0 0 Skin Cancer, Non-Melaoma 148 19 1 0 0 0 Bladder Cancer 423 45 2 0 0 0 Endometrial Cancer 218 24 0 0 0 0 Hepatobiliary Cancer 355 27 0 0 0 0 Colorectal Cancer 1007 75 0 0 0 1 Mature B-Cell Neoplasms 134 8 0 0 2 0 Non-Small Cell Lung Cancer 1668 120 0 2 1 0 Melanoma 365 23 0 0 0 0 Appendiceal Cancer 79 4 0 0 0 0 Small Cell Lung Cancer 82 4 0 0 0 0 Prostate Cancer 717 25 7 1 0 0 Histiocytosis 22 1 0 0 0 0 Salivary Gland Cancer 114 5 0 0 0 0 Thyroid Cancer 231 10 0 0 0 0 Cancer of Unknown Primary 186 8 0 0 0 0 Breast Cancer 1324 48 3 2 0 0 Adrenocortical Carcinoma 25 1 0 0 0 0 Mature T and NK Neoplasms 29 1 0 0 0 0 Renal Cell Carcinoma 361 12 0 0 0 0 Head and Neck Cancer 186 14 0 0 0 1 Pancreatic Cancer 502 69 7 2 0 0 Glioma 553 14 1 0 0 0 Soft Tissue Sarcoma 443 13 1 0 0 0 Esophagogastric Cancer 341 8 1 0 0 0 Peripheral Nervous System 80 0 2 0 0 0 Germ Cell Tumor 288 2 0 1 0 1 Ovarian Can 244 2 1 0 0 0 Uterine Sarcoma 93 1 0 0 0 0 Mesothelioma 107 1 0 0 0 0 Bone Cancer 134 1 0 0 0 0 Gastrointestinal Stromal Ca 137 0 0 0 0 1 Supplementary Table S3. -

Microarray Comparative Genomic Hybridization Analysis of 59 Patients with Schizophrenia
J Hum Genet (2008) 53:914–919 DOI 10.1007/s10038-008-0327-6 ORIGINAL ARTICLE Microarray comparative genomic hybridization analysis of 59 patients with schizophrenia Takeshi Mizuguchi Æ Ryota Hashimoto Æ Masanari Itokawa Æ Akira Sano Æ Osamu Shimokawa Æ Yukiko Yoshimura Æ Naoki Harada Æ Noriko Miyake Æ Akira Nishimura Æ Hirotomo Saitsu Æ Nadiya Sosonkina Æ Norio Niikawa Æ Hiroshi Kunugi Æ Naomichi Matsumoto Received: 18 April 2008 / Accepted: 8 July 2008 / Published online: 7 August 2008 Ó The Japan Society of Human Genetics and Springer 2008 Abstract Schizophrenia is a common psychiatric disor- that genome-wide copy number survey should be consid- der with a strong genetic contribution. Disease-associated ered in genetic studies of schizophrenia. chromosomal abnormalities in this condition may provide important clues, such as DISC1. In this study, 59 schizo- Keywords Schizophrenia Á Chromosomal abnormality Á phrenia patients were analyzed by microarray comparative Array comparative genomic hybridization Á genomic hybridization (CGH) using custom bacterial arti- Copy number variation ficial chromosome (BAC) microarray (4,219 BACs with 0.7-Mb resolution). Chromosomal abnormalities were found in six patients (10%): 46,XY,der(13)t(12;13)(p12.1; Introduction p11).ish del(5)(p11p12); 46,XY, ish del(17)(p12p12); 46,XX.ish dup(11)(p13p13); and 46,X,idic(Y)(q11.2); and Schizophrenia is a common psychiatric disorder involving in two cases, mos 45,X/46XX. Autosomal abnormalities in approximately 1% of the population worldwide. Family, three cases are likely to be pathogenic, and sex chromo- twin, and adoption studies suggest genetic factors con- some abnormalities in three follow previous findings. -

Transcriptomic and Proteomic Landscape of Mitochondrial
TOOLS AND RESOURCES Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals Inge Ku¨ hl1,2†*, Maria Miranda1†, Ilian Atanassov3, Irina Kuznetsova4,5, Yvonne Hinze3, Arnaud Mourier6, Aleksandra Filipovska4,5, Nils-Go¨ ran Larsson1,7* 1Department of Mitochondrial Biology, Max Planck Institute for Biology of Ageing, Cologne, Germany; 2Department of Cell Biology, Institute of Integrative Biology of the Cell (I2BC) UMR9198, CEA, CNRS, Univ. Paris-Sud, Universite´ Paris-Saclay, Gif- sur-Yvette, France; 3Proteomics Core Facility, Max Planck Institute for Biology of Ageing, Cologne, Germany; 4Harry Perkins Institute of Medical Research, The University of Western Australia, Nedlands, Australia; 5School of Molecular Sciences, The University of Western Australia, Crawley, Australia; 6The Centre National de la Recherche Scientifique, Institut de Biochimie et Ge´ne´tique Cellulaires, Universite´ de Bordeaux, Bordeaux, France; 7Department of Medical Biochemistry and Biophysics, Karolinska Institutet, Stockholm, Sweden Abstract Dysfunction of the oxidative phosphorylation (OXPHOS) system is a major cause of human disease and the cellular consequences are highly complex. Here, we present comparative *For correspondence: analyses of mitochondrial proteomes, cellular transcriptomes and targeted metabolomics of five [email protected] knockout mouse strains deficient in essential factors required for mitochondrial DNA gene (IKu¨ ); expression, leading to OXPHOS dysfunction. Moreover, -

Table SI. List of Differentially Expressed Genes Detected from Three Expression Profiling Datasets GSE18842, GSE30219 and GSE33532
Table SI. List of differentially expressed genes detected from three expression profiling datasets GSE18842, GSE30219 and GSE33532. DEGs Gene Upregulated (n=428) ABCB6, ACP1, ADAM12, ADAMDEC1, ADAMTS12, AFAP1-AS1, AKR1B10, ALDH3B2, ALYREF, ANKRD22, APOBEC3B, ARG2, ARNTL2, ARTN, ASF1B, ATAD2, ATP11B, AUNIP, AURKB, BAIAP2L1, BCL11A, BIK, BIRC5, BLM, BNIP3, BORA, BUB1, BYSL, C10orf2, C12orf56, C12orf66, C15orf48, C2CD4A, C4orf46, C5orf46, CABYR, CALU, CASC7, CBX2, CCNA2, CCNE1, CCNE2, CCT2, CCT6A, CDC25A, CDC25C, CDC45, CDCA2, CDCA3, CDCA8, CDH3, CDK1, CDK5R1, CDKN2A, CDT1, CEMIP, CENPF, CENPH, CENPI, CENPK, CENPL, CENPM, CENPN, MLF1IP, CENPW, CERS6, CHAC2, CHEK1, CIART, CKAP2, CKAP2L, CKAP4, CKS1B, CNFN, COCH, COL11A1, COL3A1, COL5A1, COL5A2, CPD, CRABP2, CST1, CTSV, CTTN, CXCL13, DARS2, DBF4, DCAF13, DCUN1D1, DCUN1D5, DENND1A, DEPDC1, DEPDC1B, DEPDC7, DHFR, DIO2, DONSON, DPY19L1, DSC2, DSCC1, DSG2, DST, DTL, DUS4L, E2F7, ECT2, EGLN3, EIF4EBP1, ELOVL6, ENAH, ENO1, EPT1, ERCC6L, ERO1A, ESPL1, ESRP1, ETV4, EZH2, F12, FAM169A, FAM210A, FAM64A, FAM69A, FAM83A, FAM83D, FAM83F, FAM83H, FANCD2, FANCI, FAP, FAXC, FBXO32, FBXO45, FBXO5, FERMT1, FGF11, FHL2, FLAD1, FNDC1, FOXM1, GAL, GALNT14, GALNT2, GALNT7, GART, GCLC, GCLM, GGH, GINS3, GJB2, GJB6, GMPS, GOLM1, GPX2, GRHL1, GRTP1, GTSE1, H2AFX, HELLS, HILPDA, HIST1H2AE, HIST1H2BC, HIST1H2BH, HIST1H2BJ, HIST1H3F, HIST3H2A, HLTF, HMGA1, HMGA2, HMGB3, HOOK1, HORMAD1, HOXA10, HOXC10, HOXD10, HRASLS, HS6ST2, HSPA4L, HYLS1, IDH2, IGF2BP3, IGFBP2, IGFBP3, IGHM, IGSF9, IL4I1, IQGAP3, IRF6, ITGA11, KCNK1, -

Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity
Diabetes Volume 64, September 2015 3135 Sini Heinonen,1 Jana Buzkova,2 Maheswary Muniandy,1 Risto Kaksonen,1,3 Miina Ollikainen,4 Khadeeja Ismail,4 Antti Hakkarainen,5 Jesse Lundbom,5,6 Nina Lundbom,5 Katriina Vuolteenaho,7 Eeva Moilanen,7 Jaakko Kaprio,8,9,10 Aila Rissanen,1,11 Anu Suomalainen,2,12 and Kirsi H. Pietiläinen1,8,13 Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity Diabetes 2015;64:3135–3145 | DOI: 10.2337/db14-1937 Low mitochondrial number and activity have been sug- and OXPHOS proteins in SAT are downregulated in ac- gested as underlying factors in obesity, type 2 diabetes, quired obesity, and are associated with metabolic distur- and metabolic syndrome. However, the stage at which bances already at the preclinical stage. mitochondrial dysfunction manifests in adipose tissue after the onset of obesity remains unknown. Here we examined subcutaneous adipose tissue (SAT) samples Adipocytes are important contributors to energy balance – from healthy monozygotic twin pairs, 22.8 36.2 years of and metabolic homeostasis. Within these highly dynamic D > 2 age, who were discordant ( BMI 3kg/m, mean length cells, mitochondria are at the center of energy metabo- OBESITY STUDIES 6 n of discordance 6.3 0.3 years, = 26) and concordant lism, using carbohydrates, lipids, and proteins to produce D < 2 n ( BMI 3kg/m, = 14) for body weight, and assessed ATP and metabolites for growth, as well as contributing to their detailed mitochondrial metabolic characteristics: adipocyte differentiation and maturation (1). Mitochon- mitochondrial-related transcriptomes with dysregulated dria possess their own multicopy genome, a 16.6-kb cir- pathways, mitochondrial DNA (mtDNA) amount, mtDNA- cular mitochondrial DNA (mtDNA) that encodes two encoded transcripts, and mitochondrial oxidative phos- phorylation (OXPHOS) protein levels. -
Mitochondrial Ribosomal Proteins: Candidate Genes for Mitochondrial Disease James E
March/April 2004 ⅐ Vol. 6 ⅐ No. 2 review Mitochondrial ribosomal proteins: Candidate genes for mitochondrial disease James E. Sylvester, PhD1, Nathan Fischel-Ghodsian, MD2, Edward B. Mougey, PhD1, and Thomas W. O’Brien, PhD3 Most of the energy requirement for cell growth, differentiation, and development is met by the mitochondria in the form of ATP produced by the process of oxidative phosphorylation. Human mitochondrial DNA encodes a total of 13 proteins, all of which are essential for oxidative phosphorylation. The mRNAs for these proteins are translated on mitochondrial ribosomes. Recently, the genes for human mitochondrial ribosomal proteins (MRPs) have been identified. In this review, we summarize their refined chromosomal location. It is well known that mutations in the mitochondrial translation system, i.e., ribosomal RNA and transfer RNA cause various pathologies. In this review, we suggest possible associations between clinical conditions and MRPs based on coincidence of genetic map data and chromosomal location. These MRPs may be candidate genes for the clinical condition or may act as modifiers of existing known gene mutations (mt-tRNA, mt-rRNA, etc.). Genet Med 2004:6(2):73–80. Key Words: mitochondrial, ribosomal proteins, oxidative phosphorylation, candidate genes, translation Most of the energy requirements for cell growth, differenti- THE MITORIBOSOME ation, and development are met by the mitochondrial ATP Human cells contain two genomes and two protein synthe- produced by the process of oxidative phosphorylation. Mito- sizing (translation) systems. The first is the nuclear genome of chondrial DNA encodes 13 essential proteins of this oxidative 3 ϫ 109 bp that has 30,000 to 40,000 genes coding a much phosphorylation system. -

Supplementary Information
Supplementary Information Figure S1: Normalization strategy of BioID results. Normalization of protein MS-Label Free Quantification (LFQ) values between NHA9-BioID and BirAR118G and SN214-BioID and BirAR118G. A two-step normalization approach was used to account for the differences in protein expression between the BioID fusion proteins and the control BirAR118G. 1. Internal normalization (LFQi): for each condition, the ratio between the LFQ value of each protein (protein A) and the LFQ value of the BirAR118G within the same condition (i.e. the BirA portion from the respective BioID fusion protein). 2. External normalization (LFQe): the LFQi value of each protein (protein A) in NHA9-BioID or SN214-BioID was divided by the corresponding LFQi value in the control condition (BirA). The results are expressed as a fold-change, which corresponds to log2. Figure S2: Clustered Pathway Analysis of NUP98-HOXA9-BioID proximal interactors. Functionally grouped NHA9- BioID interactors. Statistical analysis was performed with the Cytoscape plugin ClueGo (v2.5.5) using hypergeometric test and the following parameters: p <0.01, kappa-score (k) = 0.4; (min/%) genes = 3/4%, GO tree levels: 3-11. Ontology databases: GOBP, GOCC, and GOMF, Reactome Pathways and KEGG. Figure S3: Most represented cellular compartments (GOCC) among SN214-BioID proximal interactors. Statistical analysis of the overrepresented proteins in the SN214-BioID fraction (total 1125 proteins) with PANTHER classification online software (v14.1) using Fisher’s exact test. Results are displayed for FDR p <0.05. Figure S4: Most represented biological processes (GOBP) among SN214-BioID proximal interactors. Statistical analysis of the overrepresented proteins in the SN214-BioID fraction (total 1125 proteins) with PANTHER classification online software (v14.1) using Fisher’s exact test. -
Table 1 the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table 1 The statistical metrics for key differentially expressed genes (DEGs) Illumina Id Gene Symbol logFC p Value FDR t value Regulation Gene name ILMN_1757497 VGF 4.848711 1.27E-13 6.11E-09 48.36804 UP VGF nerve growth factor inducible ILMN_2193706 HRK 3.058491 2.89E-10 3.47E-06 22.91096 UP harakiri, BCL2 interacting protein ILMN_2182198 MRPL58 1.8013 5.14E-10 3.64E-06 21.65441 UP mitochondrial ribosomal protein L58 ILMN_1699772 RRAGD 2.331637 6.56E-10 3.64E-06 21.14388 UP Ras related GTP binding D ILMN_1695969 TMPRSS15 2.158671 6.81E-10 3.64E-06 21.06686 UP transmembrane protease, serine 15 ILMN_1754002 IL36B 4.161387 3.21E-09 1.10E-05 18.09187 UP interleukin 36, beta ILMN_1656134 CNOT7 1.387716 4.97E-09 1.49E-05 17.32692 UP CCR4-NOT transcription complex subunit 7 ILMN_1658499 SYT13 1.653015 6.80E-09 1.59E-05 16.79842 UP synaptotagmin 13 ILMN_1684836 AKAP12 1.291595 8.20E-09 1.68E-05 16.48925 UP A-kinase anchoring protein 12 ILMN_1799519 IL36B 4.299495 9.58E-09 1.68E-05 16.23691 UP interleukin 36, beta ILMN_1678757 BCYRN1 3.056229 9.58E-09 1.68E-05 16.23661 UP brain cytoplasmic RNA 1 ILMN_1726743 MRPS30 1.302467 1.08E-08 1.73E-05 16.04765 UP mitochondrial ribosomal protein S30 ILMN_1777976 SLC25A26 1.609861 1.37E-08 2.06E-05 15.66752 UP solute carrier family 25 member 26 mitochondrially encoded ILMN_3308961 MT-ND6 3.019664 1.61E-08 2.15E-05 15.41788 UP NADH:ubiquinoneoxidoreductase core subunit 6 ILMN_2380999 RECQL 1.437639 2.36E-08 2.72E-05 14.83968 UP RecQ like helicase ILMN_3233229 SNHG7 1.202524 2.50E-08 2.72E-05 14.7569 UP small -

Research Collection
Research Collection Journal Article The complete structure of the large subunit of the mammalian mitochondrial ribosome Author(s): Greber, Basil J.; Boehringer, Daniel; Leibundgut, Marc; Bieri, Philipp; Leitner, Alexander; Schmitz, Nikolaus; Aebersold, Ruedi; Ban, Nenad Publication Date: 2014-10-01 Permanent Link: https://doi.org/10.3929/ethz-b-000093239 Originally published in: Nature 515(7526), http://doi.org/10.1038/nature13895 Rights / License: In Copyright - Non-Commercial Use Permitted This page was generated automatically upon download from the ETH Zurich Research Collection. For more information please consult the Terms of use. ETH Library Supplementary Online Material for The Complete Structure of the Large Subunit of the Mammalian Mitoribosome Basil J. Greber*, Daniel Boehringer*, Marc Leibundgut*, Philipp Bieri, Alexander Leitner, Nikolaus Schmitz, Ruedi Aebersold, and Nenad Ban† * These authors contributed equally to this work † Correspondence to: [email protected] (NB) This file contains: Supplementary Discussion Supplementary Table 1 Supplementary Information References 59-69 Provided in a dedicated spreadsheet file: Supplementary Table 2 Supplementary Discussion Identification of mL65 (MRPS30) and mL66 (MRPS18A) as 39S Subunit Components The mammalian mitochondrial ribosomal specific proteins mL37 and mL65 (MRPS30; previously termed PDCD9 59 or p52 60) are homologous to each other (Extended Data Fig. 5b, c) and dimerize via an extensive interface (1430 Å2; computed with PDBePISA 61). The presence of mL65 (MRPS30) on the 39S subunit is unexpected, as it was initially identified as a protein of the small mitoribosomal subunit 59. However, our assignment based on the inspection of the well-resolved amino acid side chains in the cryo-EM density was unambiguous (Extended Data Fig.