Study of the Pathology and the Underlying Molecular Mechanism of Tissue Injury Around Hematoma Following Intracerebral Hemorrhage
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Data Sheet 160-3P
Rudolf-Wissell-Str. 28a Background 37079 Göttingen, Germany Phone: +49 551-50556-0 Homer is a scaffolding protein of the post synaptic density (PSD) and enriched at excitatory synapses. Fax: +49 551-50556-384 The protein binds metabotropic glutamate receptors, TRPC1, proteins of the Shank family and others. E-mail: [email protected] By aggregating these proteins into clusters, Homer was suggested to organize distinct signalling Web: www.sysy.com domains. Homer 3 Three isoforms, Homer 1, 2 and 3 have been described. Each of these isoforms is subject to alternative splicing yielding the splice variants a, b, c, d. Cat.No. 160-3P; control protein, 100 µg protein (lyophilized) Data Sheet Selected General References Homer2 and Homer3 interact with amyloid precursor protein and inhibit Abeta production. Parisiadou L, Bethani I, Michaki V, Krousti K, Rapti G, Efthimiopoulos S Reconstitution/ 100 µg lyophilized protein. For reconstitution add 100 µl H2O to get a 1mg/ml Neurobiology of disease (2008) 303: 353-64. Storage solution in MBS. Then aliquot and store at -20°C until use. Differential expression of Homer family proteins in the developing mouse brain. For detailed information, see back of the data sheet. Shiraishi Y, Mizutani A, Yuasa S, Mikoshiba K, Furuichi T The Journal of comparative neurology (2004) 4734: 582-99. Immunogen Recombinant protein corresponding to AA 1 to 177 from rat Homer3 (UniProt Id: Q9Z2X5) Molecular characterisation of two structurally distinct groups of human homers, generated by extensive alternative splicing. Soloviev MM, Ciruela F, Chan WY, McIlhinney RA Recommended Optimal concentrations should be determined by the end-user. -

Noelia Díaz Blanco
Effects of environmental factors on the gonadal transcriptome of European sea bass (Dicentrarchus labrax), juvenile growth and sex ratios Noelia Díaz Blanco Ph.D. thesis 2014 Submitted in partial fulfillment of the requirements for the Ph.D. degree from the Universitat Pompeu Fabra (UPF). This work has been carried out at the Group of Biology of Reproduction (GBR), at the Department of Renewable Marine Resources of the Institute of Marine Sciences (ICM-CSIC). Thesis supervisor: Dr. Francesc Piferrer Professor d’Investigació Institut de Ciències del Mar (ICM-CSIC) i ii A mis padres A Xavi iii iv Acknowledgements This thesis has been made possible by the support of many people who in one way or another, many times unknowingly, gave me the strength to overcome this "long and winding road". First of all, I would like to thank my supervisor, Dr. Francesc Piferrer, for his patience, guidance and wise advice throughout all this Ph.D. experience. But above all, for the trust he placed on me almost seven years ago when he offered me the opportunity to be part of his team. Thanks also for teaching me how to question always everything, for sharing with me your enthusiasm for science and for giving me the opportunity of learning from you by participating in many projects, collaborations and scientific meetings. I am also thankful to my colleagues (former and present Group of Biology of Reproduction members) for your support and encouragement throughout this journey. To the “exGBRs”, thanks for helping me with my first steps into this world. Working as an undergrad with you Dr. -
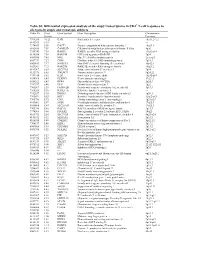
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3 -

Statistical and Bioinformatic Analysis of Hemimethylation Patterns in Non-Small Cell Lung Cancer
Statistical and Bioinformatic Analysis of Hemimethylation Patterns in Non-Small Cell Lung Cancer Shuying Sun ( [email protected] ) Texas State University San Marcos https://orcid.org/0000-0003-3974-6996 Austin Zane Texas A&M University College Station Carolyn Fulton Schreiner University Jasmine Philipoom Case Western Reserve University Research article Keywords: Methylation, Hemimethylation, Lung Cancer, Bioinformatics, Epigenetics Posted Date: October 12th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-17794/v2 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published on March 12th, 2021. See the published version at https://doi.org/10.1186/s12885-021-07990-7. Page 1/29 Abstract Background: DNA methylation is an epigenetic event involving the addition of a methyl-group to a cytosine-guanine base pair (i.e., CpG site). It is associated with different cancers. Our research focuses on studying non- small cell lung cancer hemimethylation, which refers to methylation occurring on only one of the two DNA strands. Many studies often assume that methylation occurs on both DNA strands at a CpG site. However, recent publications show the existence of hemimethylation and its signicant impact. Therefore, it is important to identify cancer hemimethylation patterns. Methods: In this paper, we use the Wilcoxon signed rank test to identify hemimethylated CpG sites based on publicly available non-small cell lung cancer methylation sequencing data. We then identify two types of hemimethylated CpG clusters, regular and polarity clusters, and genes with large numbers of hemimethylated sites. -

Annual Scientific Report 2013 on the Cover Structure 3Fof in the Protein Data Bank, Determined by Laponogov, I
EMBL-European Bioinformatics Institute Annual Scientific Report 2013 On the cover Structure 3fof in the Protein Data Bank, determined by Laponogov, I. et al. (2009) Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nature Structural & Molecular Biology 16, 667-669. © 2014 European Molecular Biology Laboratory This publication was produced by the External Relations team at the European Bioinformatics Institute (EMBL-EBI) A digital version of the brochure can be found at www.ebi.ac.uk/about/brochures For more information about EMBL-EBI please contact: [email protected] Contents Introduction & overview 3 Services 8 Genes, genomes and variation 8 Molecular atlas 12 Proteins and protein families 14 Molecular and cellular structures 18 Chemical biology 20 Molecular systems 22 Cross-domain tools and resources 24 Research 26 Support 32 ELIXIR 36 Facts and figures 38 Funding & resource allocation 38 Growth of core resources 40 Collaborations 42 Our staff in 2013 44 Scientific advisory committees 46 Major database collaborations 50 Publications 52 Organisation of EMBL-EBI leadership 61 2013 EMBL-EBI Annual Scientific Report 1 Foreword Welcome to EMBL-EBI’s 2013 Annual Scientific Report. Here we look back on our major achievements during the year, reflecting on the delivery of our world-class services, research, training, industry collaboration and European coordination of life-science data. The past year has been one full of exciting changes, both scientifically and organisationally. We unveiled a new website that helps users explore our resources more seamlessly, saw the publication of ground-breaking work in data storage and synthetic biology, joined the global alliance for global health, built important new relationships with our partners in industry and celebrated the launch of ELIXIR. -

Brain Region-Dependent Gene Networks Associated with Selective Breeding for Increased Voluntary Wheel-Running Behavior
UC Riverside UC Riverside Previously Published Works Title Brain region-dependent gene networks associated with selective breeding for increased voluntary wheel-running behavior. Permalink https://escholarship.org/uc/item/8c49g8fd Journal PloS one, 13(8) ISSN 1932-6203 Authors Zhang, Pan Rhodes, Justin S Garland, Theodore et al. Publication Date 2018 DOI 10.1371/journal.pone.0201773 Peer reviewed eScholarship.org Powered by the California Digital Library University of California RESEARCH ARTICLE Brain region-dependent gene networks associated with selective breeding for increased voluntary wheel-running behavior Pan Zhang1,2, Justin S. Rhodes3,4, Theodore Garland, Jr.5, Sam D. Perez3, Bruce R. Southey2, Sandra L. Rodriguez-Zas2,6,7* 1 Illinois Informatics Institute, University of Illinois at Urbana-Champaign, Urbana, IL, United States of America, 2 Department of Animal Sciences, University of Illinois at Urbana-Champaign, Urbana, IL, United a1111111111 States of America, 3 Beckman Institute for Advanced Science and Technology, Urbana, IL, United States of a1111111111 America, 4 Center for Nutrition, Learning and Memory, University of Illinois at Urbana-Champaign, Urbana, a1111111111 IL, United States of America, 5 Department of Evolution, Ecology, and Organismal Biology, University of a1111111111 California, Riverside, CA, United States of America, 6 Department of Statistics, University of Illinois at Urbana-Champaign, Urbana, IL, United States of America, 7 Carle Woese Institute for Genomic Biology, a1111111111 University of Illinois at Urbana-Champaign, Urbana, IL, United States of America * [email protected] OPEN ACCESS Abstract Citation: Zhang P, Rhodes JS, Garland T, Jr., Perez SD, Southey BR, Rodriguez-Zas SL (2018) Brain Mouse lines selectively bred for high voluntary wheel-running behavior are helpful models region-dependent gene networks associated with for uncovering gene networks associated with increased motivation for physical activity and selective breeding for increased voluntary wheel- other reward-dependent behaviors. -

X-Linked Diseases: Susceptible Females
REVIEW ARTICLE X-linked diseases: susceptible females Barbara R. Migeon, MD 1 The role of X-inactivation is often ignored as a prime cause of sex data include reasons why women are often protected from the differences in disease. Yet, the way males and females express their deleterious variants carried on their X chromosome, and the factors X-linked genes has a major role in the dissimilar phenotypes that that render women susceptible in some instances. underlie many rare and common disorders, such as intellectual deficiency, epilepsy, congenital abnormalities, and diseases of the Genetics in Medicine (2020) 22:1156–1174; https://doi.org/10.1038/s41436- heart, blood, skin, muscle, and bones. Summarized here are many 020-0779-4 examples of the different presentations in males and females. Other INTRODUCTION SEX DIFFERENCES ARE DUE TO X-INACTIVATION Sex differences in human disease are usually attributed to The sex differences in the effect of X-linked pathologic variants sex specific life experiences, and sex hormones that is due to our method of X chromosome dosage compensation, influence the function of susceptible genes throughout the called X-inactivation;9 humans and most placental mammals – genome.1 5 Such factors do account for some dissimilarities. compensate for the sex difference in number of X chromosomes However, a major cause of sex-determined expression of (that is, XX females versus XY males) by transcribing only one disease has to do with differences in how males and females of the two female X chromosomes. X-inactivation silences all X transcribe their gene-rich human X chromosomes, which is chromosomes but one; therefore, both males and females have a often underappreciated as a cause of sex differences in single active X.10,11 disease.6 Males are the usual ones affected by X-linked For 46 XY males, that X is the only one they have; it always pathogenic variants.6 Females are biologically superior; a comes from their mother, as fathers contribute their Y female usually has no disease, or much less severe disease chromosome. -

SHOC2–MRAS–PP1 Complex Positively Regulates RAF Activity and Contributes to Noonan Syndrome Pathogenesis
SHOC2–MRAS–PP1 complex positively regulates RAF activity and contributes to Noonan syndrome pathogenesis Lucy C. Younga,1, Nicole Hartiga,2, Isabel Boned del Ríoa, Sibel Saria, Benjamin Ringham-Terrya, Joshua R. Wainwrighta, Greg G. Jonesa, Frank McCormickb,3, and Pablo Rodriguez-Vicianaa,3 aUniversity College London Cancer Institute, University College London, London WC1E 6DD, United Kingdom; and bHelen Diller Family Comprehensive Cancer Center, University of California, San Francisco, CA 94158 Contributed by Frank McCormick, September 18, 2018 (sent for review November 22, 2017; reviewed by Deborah K. Morrison and Marc Therrien) Dephosphorylation of the inhibitory “S259” site on RAF kinases CRAF/RAF1 mutations are also frequently found in NS and (S259 on CRAF, S365 on BRAF) plays a key role in RAF activation. cluster around the S259 14-3-3 binding site, enhancing CRAF ac- The MRAS GTPase, a close relative of RAS oncoproteins, interacts tivity through disruption of 14-3-3 binding (8) and highlighting the with SHOC2 and protein phosphatase 1 (PP1) to form a heterotri- key role of this regulatory step in RAF–ERK pathway activation. meric holoenzyme that dephosphorylates this S259 RAF site. MRAS is a very close relative of the classical RAS oncoproteins MRAS and SHOC2 function as PP1 regulatory subunits providing (H-, N-, and KRAS, hereafter referred to collectively as “RAS”) the complex with striking specificity against RAF. MRAS also func- and shares most regulatory and effector interactions as well as tions as a targeting subunit as membrane localization is required transforming ability (9–11). However, MRAS also has specific for efficient RAF dephosphorylation and ERK pathway regulation functions of its own, and uniquely among RAS family GTPases, it in cells. -

Decoding DMD Transcriptional Networks Using Single‐Nucleus RNA Sequencing COMMENTARY Daniel J
COMMENTARY Decoding DMD transcriptional networks using single‐nucleus RNA sequencing COMMENTARY Daniel J. Garrya,b,1, Satyabrata Dasa, and Wuming Gonga Duchenne muscular dystrophy (DMD) is an X chromosome-linked disease, and it is the most com- mon form of muscular dystrophy caused by genetic mutations in the Dmd gene (1). The Dmd gene contains 79 exons, spans 2.4 Mb, and is the single largest gene in the human genome (2). This gene encodes for dystro- phin, which is a component of the dystrophin– glycoprotein complex that provides structural stability to the cell membrane (by connecting the cytoskeleton and the extracellular matrix), and a dys- functional or absent dystrophin protein leads to pro- Fig. 1. Decoding the molecular mechanisms of DMD gressive muscle wasting with cycles of muscle pathogenesis utilizing snRNA-seq. This figure schematizes the discoveries outlined in Chemello et al. degeneration and regeneration that ultimately fail (3, (7) with the newly generated DMD mouse model 4). Young boys with DMD lose their ability to ambu- (ΔEx51) and the transcriptomic analysis of isolated late, become wheelchair bound, and die prematurely nuclei from skeletal muscle cells. The snRNA-seq (1, 5). This disease affects skeletal muscle, which is the analysis was used to discover factors and pathways underlying DMD pathogenesis from previously single largest organ in the body, and it normally has a unrecognized cell clusters. These results in remarkable capacity for regeneration (6). In response combination with other databases provide a platform to a severe injury caused by genetic disorders, trauma, for developing new therapeutic interventions or exposure to toxins that destroys over 90% of the mus- as well as an enhanced understanding of the gene-regulatory networks involved in muscle biology cle, the cytoarchitecture of the injured tissue is com- and DMD. -

The Landscape of Human Mutually Exclusive Splicing
bioRxiv preprint doi: https://doi.org/10.1101/133215; this version posted May 2, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. The landscape of human mutually exclusive splicing Klas Hatje1,2,#,*, Ramon O. Vidal2,*, Raza-Ur Rahman2, Dominic Simm1,3, Björn Hammesfahr1,$, Orr Shomroni2, Stefan Bonn2§ & Martin Kollmar1§ 1 Group of Systems Biology of Motor Proteins, Department of NMR-based Structural Biology, Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germany 2 Group of Computational Systems Biology, German Center for Neurodegenerative Diseases, Göttingen, Germany 3 Theoretical Computer Science and Algorithmic Methods, Institute of Computer Science, Georg-August-University Göttingen, Germany § Corresponding authors # Current address: Roche Pharmaceutical Research and Early Development, Pharmaceutical Sciences, Roche Innovation Center Basel, F. Hoffmann-La Roche Ltd., Basel, Switzerland $ Current address: Research and Development - Data Management (RD-DM), KWS SAAT SE, Einbeck, Germany * These authors contributed equally E-mail addresses: KH: [email protected], RV: [email protected], RR: [email protected], DS: [email protected], BH: [email protected], OS: [email protected], SB: [email protected], MK: [email protected] - 1 - bioRxiv preprint doi: https://doi.org/10.1101/133215; this version posted May 2, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Roles of Nickel Binding Proteins in Helicobacter Species
ROLES OF NICKEL BINDING PROTEINS IN HELICOBACTER SPECIES by SUSMITHA SESHADRI (Under the Direction of Robert J. Maier) ABSTRACT Proteins Hpn and Hpn-like of the gastric pathogen H. pylori were hypothesized to bind nickel since histidine residues make up 45% and 27% of their amino acid content, respectively. Characterization of an hpn, an hpn-like and an hpn, hpn-like double mutant revealed novel functions for these gene products in nickel detoxification and storage. Compared to the wild-type parent, mutant strains were more sensitive to elevated concentrations of nickel, cobalt, and cadmium, indicating roles for the two proteins in surviving metal toxicity. Under low nickel conditions, the mutants exhibited higher urease activities and had increased amount of Ni- associated with urease; but similar urease apo-protein levels to wild-type. The parent achieved mutant level urease activities under nickel supplementation and lower pH conditions while growth with a nickel chelator decreased mutant but not wild-type urease activities. These results strongly imply a role for these proteins as nickel reservoirs/storage proteins. H. hepaticus colonizes a non-acidic niche, hence nickel metabolism may be different than in H. pylori. A nikR mutant exhibited higher urease and hydrogenase activities under all supplemental nickel conditions, but there was no change in urease expression, and NikR did not bind pUreA or pHydA. Higher total nickel levels (detected by ICP-MS) in the nikR mutant implied possible higher nickel transporter (NikA) levels, which was later verified by qRT-PCR and binding of NikR to pNikA. Periplasmic nitrate reductase (NapA) was upregulated in the NikR strain. -

(12) United States Patent (10) Patent No.: US 7.873,482 B2 Stefanon Et Al
US007873482B2 (12) United States Patent (10) Patent No.: US 7.873,482 B2 Stefanon et al. (45) Date of Patent: Jan. 18, 2011 (54) DIAGNOSTIC SYSTEM FOR SELECTING 6,358,546 B1 3/2002 Bebiak et al. NUTRITION AND PHARMACOLOGICAL 6,493,641 B1 12/2002 Singh et al. PRODUCTS FOR ANIMALS 6,537,213 B2 3/2003 Dodds (76) Inventors: Bruno Stefanon, via Zilli, 51/A/3, Martignacco (IT) 33035: W. Jean Dodds, 938 Stanford St., Santa Monica, (Continued) CA (US) 90403 FOREIGN PATENT DOCUMENTS (*) Notice: Subject to any disclaimer, the term of this patent is extended or adjusted under 35 WO WO99-67642 A2 12/1999 U.S.C. 154(b) by 158 days. (21)21) Appl. NoNo.: 12/316,8249 (Continued) (65) Prior Publication Data Swanson, et al., “Nutritional Genomics: Implication for Companion Animals'. The American Society for Nutritional Sciences, (2003).J. US 2010/O15301.6 A1 Jun. 17, 2010 Nutr. 133:3033-3040 (18 pages). (51) Int. Cl. (Continued) G06F 9/00 (2006.01) (52) U.S. Cl. ........................................................ 702/19 Primary Examiner—Edward Raymond (58) Field of Classification Search ................... 702/19 (74) Attorney, Agent, or Firm Greenberg Traurig, LLP 702/23, 182–185 See application file for complete search history. (57) ABSTRACT (56) References Cited An analysis of the profile of a non-human animal comprises: U.S. PATENT DOCUMENTS a) providing a genotypic database to the species of the non 3,995,019 A 1 1/1976 Jerome human animal Subject or a selected group of the species; b) 5,691,157 A 1 1/1997 Gong et al.