7 Natural Abundance of the Stable Isotopes of C, O and H
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

THE NATURAL RADIOACTIVITY of the BIOSPHERE (Prirodnaya Radioaktivnost' Iosfery)
XA04N2887 INIS-XA-N--259 L.A. Pertsov TRANSLATED FROM RUSSIAN Published for the U.S. Atomic Energy Commission and the National Science Foundation, Washington, D.C. by the Israel Program for Scientific Translations L. A. PERTSOV THE NATURAL RADIOACTIVITY OF THE BIOSPHERE (Prirodnaya Radioaktivnost' iosfery) Atomizdat NMoskva 1964 Translated from Russian Israel Program for Scientific Translations Jerusalem 1967 18 02 AEC-tr- 6714 Published Pursuant to an Agreement with THE U. S. ATOMIC ENERGY COMMISSION and THE NATIONAL SCIENCE FOUNDATION, WASHINGTON, D. C. Copyright (D 1967 Israel Program for scientific Translations Ltd. IPST Cat. No. 1802 Translated and Edited by IPST Staff Printed in Jerusalem by S. Monison Available from the U.S. DEPARTMENT OF COMMERCE Clearinghouse for Federal Scientific and Technical Information Springfield, Va. 22151 VI/ Table of Contents Introduction .1..................... Bibliography ...................................... 5 Chapter 1. GENESIS OF THE NATURAL RADIOACTIVITY OF THE BIOSPHERE ......................... 6 § Some historical problems...................... 6 § 2. Formation of natural radioactive isotopes of the earth ..... 7 §3. Radioactive isotope creation by cosmic radiation. ....... 11 §4. Distribution of radioactive isotopes in the earth ........ 12 § 5. The spread of radioactive isotopes over the earth's surface. ................................. 16 § 6. The cycle of natural radioactive isotopes in the biosphere. ................................ 18 Bibliography ................ .................. 22 Chapter 2. PHYSICAL AND BIOCHEMICAL PROPERTIES OF NATURAL RADIOACTIVE ISOTOPES. ........... 24 § 1. The contribution of individual radioactive isotopes to the total radioactivity of the biosphere. ............... 24 § 2. Properties of radioactive isotopes not belonging to radio- active families . ............ I............ 27 § 3. Properties of radioactive isotopes of the radioactive families. ................................ 38 § 4. Properties of radioactive isotopes of rare-earth elements . -

The Dole Effect and Its Variations During the Last 130,000 Years As Measured in the Vostok Ice Core
GLOBAL BIOGEOCHEMICAL CYCLES, VOL. 8, NO. 3, PAGES 363-376, SEPTEMBER 1994 The Dole effect and its variations during the last 130,000 years as measured in the Vostok ice core Michael Bender • and Todd Sowers 2 GraduateSchool of Oceanography,University of RhodeIsland, Kingston Laurent Labeyrie Centredes Faibles Radioactivites, Centre National de la RechercheScientifique, Commissariat a L'EnergieAtomique, Gif-sur- Yvette, France Abstract. We review the currentunderstanding of the Dole effect (the observeddifference between the •5180of atmospheric0 2 andthat of seawater)and its causes, extend the record of variationsin theDole effectback to 130kyr before present using data on the •5180 of 0 2obtained from studying the Vostok ice core(Sowers et al., 1993), anddiscuss the significanceof temporalvariations. The Dole effectreflects oxygenisotope fractionation during photosynthesis, respiration, and hydrologic processes (evaporation, precipitation,and evapotranspiration). Our bestprediction of the present-dayDole effect,+20.8 %0,is considerablylower thanthe observedvalue, + 23.5 %0,and we discusspossible causes of this discrepancy. During the past 130 kyr, the Dole effecthas been 0.05 %0lower thanthe present value, on average.The standarddeviation of the Dole effect from the meanhas been only _+0.2 %0,and the Dole effect is nearly unchangedbetween glacial maxima and interglacial periods. The smallvariability in theDole effect suggeststhat relative rates of primaryproduction in theland and marine realms have been relatively constant.Most periodicvariability in the Dole effectis in the precessionband, suggesting that changes in thisglobal biogeochemical term reflects variations in low-latitudeland hydrology and productivity or possiblyvariability in low-latitudeoceanic productivity. Introduction they can potentially be made to do so. Ice core reconstructionsof the concentrationsof bioactivegases in air Our understanding of the response of the biosphere to provide an integratedglobal signal which complementsthe Pleistoceneclimate change is actually quite limited. -

Chemical and Isotropic Tracers of Natural Gas and Formation Waters in Fractured Shales
Chemical and isotopic tracers of natural gas and formation waters in Note: this document may contain some elements that are not fully fractured shales accessible to users with disabilities. If you need assistance accessing any information in this document, please contact [email protected]. Jennifer McIntosh, Melissa Schlegel, Brittney Bates Department of Hydrology & Water Resources University of Arizona, Tucson AZ EPA Technical Workshop Feb 24-25, 2011 Outline of Presentation 1)) What is the chemical and isotoppgic signature of formation waters and natural gas in fractured shales? 2) How does it compare with shallow drift aquifers, coalbeds, and other deep geologic formations? Illinois Basin-Case Study - 3 organic-rich formations: glacial drift, Penn. coal, & Dev. fractured shale - organicorganic-rich Ordov. ShaleShale, not part ooff this study m 500 20 km Schlegel et al. (in press, 2011, Geochimica et Cosmochimica Acta) Illinois Basin-Case Study - microbial methane in all 3 units - thermogenic methane in shale and coal m 500 20 km Schlegel et al. (in press, 2011, Geochimica et Cosmochimica Acta) Illinois Basin - water & gas samples Shallow aquifers Penn. Coals Dev. Shale Limestone aquifers 9New data (white symbols): Schlegel et al. (in press) 9Previous data (black symbols): McIntosh et al., 2002; Strąpoć et al., 2007, 2008a,b; Coleman et al, 1988 Fingerprint of natural gases 1000000 Shallow Aquifers 100000 High C1/C2, no C3 Coals 10000 1000 Shale 100 10 Shallow aquifers Penn. Coals Dev. Shale Thermogenic gas 1 -80 -75 -70 -65 -60 -55 -50 -45 -40 13 δ CCH4 (ä PDB) 13 9In general, 3 organic-rich units have different gas compositions (C1/C2+C3) and δ C-CH4 values. -

F:\Forensic Geology\Ice Man Isotopes.Wpd
NAME 89.215 - FORENSIC GEOLOGY DEMISE OF THE ICE MAN - ISOTOPIC EVIDENCE I. Introduction Stable and radiogenic isotopic data have been used in a variety of fields to answer a wide range of scientific questions. The nucleus of an atom consists of protons (+1 charge) and neutrons (0 charge), two types of particles that have essentially the same atomic mass. The number of protons in a nucleus determines the element. For example, a nucleus with 1 proton is a hydrogen nucleus, a nucleus with 2 protons is a helium nucleus. Isotopes of an element contain the same number of protons but different numbers of neutrons. For example, there are three isotopes of hydrogen: (1) ordinary hydrogen which contains one proton and no neutrons and has an atomic mass of one; (2) deuterium which contains one proton and one neutron and has an atomic mass of two; and (3) tritium which contains one proton and two neutrons and has an atomic mass of three. The convention used to show the numbers of types of particles in a nucleus is to place the number of protons (called the atomic number) at the bottom left of the element symbol and the number of protons plus neutrons (called the atomic mass) at the upper left of the element symbol. For example, the tritium isotope is 3 shown as 1H . II. Stable isotopes Stable isotopes are not radioactive, they do not spontaneously breakdown to other atoms. In a previous exercise you used radioactive carbon to determine when the Ice Man was killed. There are three isotopes of carbon: (1) carbon 12 which contains 6 protons and 6 neutrons giving an atomic mass of 12; (2) carbon 13 which contains 6 protons and 7 neutrons giving an atomic mass of 13; and (3) carbon 14 which contains 6 protons and 8 neutrons giving an atomic mass of 14. -

12 Natural Isotopes of Elements Other Than H, C, O
12 NATURAL ISOTOPES OF ELEMENTS OTHER THAN H, C, O In this chapter we are dealing with the less common applications of natural isotopes. Our discussions will be restricted to their origin and isotopic abundances and the main characteristics. Only brief indications are given about possible applications. More details are presented in the other volumes of this series. A few isotopes are mentioned only briefly, as they are of little relevance to water studies. Based on their half-life, the isotopes concerned can be subdivided: 1) stable isotopes of some elements (He, Li, B, N, S, Cl), of which the abundance variations point to certain geochemical and hydrogeological processes, and which can be applied as tracers in the hydrological systems, 2) radioactive isotopes with half-lives exceeding the age of the universe (232Th, 235U, 238U), 3) radioactive isotopes with shorter half-lives, mainly daughter nuclides of the previous catagory of isotopes, 4) radioactive isotopes with shorter half-lives that are of cosmogenic origin, i.e. that are being produced in the atmosphere by interactions of cosmic radiation particles with atmospheric molecules (7Be, 10Be, 26Al, 32Si, 36Cl, 36Ar, 39Ar, 81Kr, 85Kr, 129I) (Lal and Peters, 1967). The isotopes can also be distinguished by their chemical characteristics: 1) the isotopes of noble gases (He, Ar, Kr) play an important role, because of their solubility in water and because of their chemically inert and thus conservative character. Table 12.1 gives the solubility values in water (data from Benson and Krause, 1976); the table also contains the atmospheric concentrations (Andrews, 1992: error in his Eq.4, where Ti/(T1) should read (Ti/T)1); 2) another category consists of the isotopes of elements that are only slightly soluble and have very low concentrations in water under moderate conditions (Be, Al). -

Compilation of Minimum and Maximum Isotope Ratios of Selected Elements in Naturally Occurring Terrestrial Materials and Reagents by T
U.S. Department of the Interior U.S. Geological Survey Compilation of Minimum and Maximum Isotope Ratios of Selected Elements in Naturally Occurring Terrestrial Materials and Reagents by T. B. Coplen', J. A. Hopple', J. K. Bohlke', H. S. Reiser', S. E. Rieder', H. R. o *< A R 1 fi Krouse , K. J. R. Rosman , T. Ding , R. D. Vocke, Jr., K. M. Revesz, A. Lamberty, P. Taylor, and P. De Bievre6 'U.S. Geological Survey, 431 National Center, Reston, Virginia 20192, USA 2The University of Calgary, Calgary, Alberta T2N 1N4, Canada 3Curtin University of Technology, Perth, Western Australia, 6001, Australia Institute of Mineral Deposits, Chinese Academy of Geological Sciences, Beijing, 100037, China National Institute of Standards and Technology, 100 Bureau Drive, Stop 8391, Gaithersburg, Maryland 20899 "institute for Reference Materials and Measurements, Commission of the European Communities Joint Research Centre, B-2440 Geel, Belgium Water-Resources Investigations Report 01-4222 Reston, Virginia 2002 U.S. Department of the Interior GALE A. NORTON, Secretary U.S. Geological Survey Charles G. Groat, Director The use of trade, brand, or product names in this report is for identification purposes only and does not imply endorsement by the U.S. Government. For additional information contact: Chief, Isotope Fractionation Project U.S. Geological Survey Mail Stop 431 - National Center Reston, Virginia 20192 Copies of this report can be purchased from: U.S. Geological Survey Branch of Information Services Box 25286 Denver, CO 80225-0286 CONTENTS Abstract...................................................... -
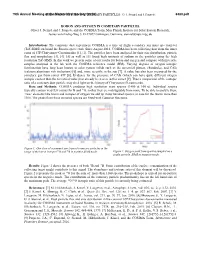
Boron and Oxygen in Cometary Particles: O. J
79th Annual Meeting ofBORON the Meteoritical AND OXYGEN Society IN COMETARY (2016) PARTICLES: O. J. Stenzel and J. Paquette 6380.pdf BORON AND OXYGEN IN COMETARY PARTICLES. Oliver J. Stenzel and J. Paquette and the COSIMA Team, Max Planck Institute for Solar System Research, Justus-von-Liebig-Weg 3, D-37077 Göttingen, Germany, [email protected]. Introduction: The cometary dust experiment COSIMA is a time of flight secondary ion mass spectrometer (ToF-SIMS) on board the Rosetta space craft. Since August 2014, COSIMA has been collecting dust from the inner coma of 67P/Churyumov-Gerasimenko [1], [2]. The particles have been analyzed for their size distribution, particle flux and morphology [3], [4]. [5] as well as [1] found high amounts of sodium in the particles using the high resolution ToF-SIMS. In this work we present some of our results for boron and oxygen and compare with meteorite samples analyzed in the lab with the COSIMA reference model (RM). Varying degrees of oxygen isotopic fractionation have long been known in solar system solids such as the terrestrial planets, chondrules, and CAIs (calcium-aluminum-rich inclusions) [6] and, more recently, in the sun [7]. A value has also been measured for the cometary gas from comet 67P [8]. Evidence for the presence of CAIs (which can have quite different oxygen isotopic content than the terrestrial value) has already been seen in this comet [9]. Thus a comparison of the isotopic ratio of a cometary dust particle may shed light on the history of Churyumov-Gerasimenko. Data and Methods: COSIMA produces high resolution mass spectra (1400 at 100 u). -

Investigation of Nitrogen Cycling Using Stable Nitrogen and Oxygen Isotopes in Narragansett Bay, Ri
University of Rhode Island DigitalCommons@URI Open Access Dissertations 2014 INVESTIGATION OF NITROGEN CYCLING USING STABLE NITROGEN AND OXYGEN ISOTOPES IN NARRAGANSETT BAY, RI Courtney Elizabeth Schmidt University of Rhode Island, [email protected] Follow this and additional works at: https://digitalcommons.uri.edu/oa_diss Recommended Citation Schmidt, Courtney Elizabeth, "INVESTIGATION OF NITROGEN CYCLING USING STABLE NITROGEN AND OXYGEN ISOTOPES IN NARRAGANSETT BAY, RI" (2014). Open Access Dissertations. Paper 209. https://digitalcommons.uri.edu/oa_diss/209 This Dissertation is brought to you for free and open access by DigitalCommons@URI. It has been accepted for inclusion in Open Access Dissertations by an authorized administrator of DigitalCommons@URI. For more information, please contact [email protected]. INVESTIGATION OF NITROGEN CYCLING USING STABLE NITROGEN AND OXYGEN ISOTOPES IN NARRAGANSETT BAY, RI BY COURTNEY ELIZABETH SCHMIDT A DISSERTATION SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN OCEANOGRAPHY UNIVERSITY OF RHODE ISLAND 2014 DOCTOR OF PHILOSOPHY DISSERTATION OF COURTNEY ELIZABETH SCHMIDT APPROVED: Thesis Committee: Major Professor Rebecca Robinson Candace Oviatt Arthur Gold Nassar H. Zawia DEAN OF THE GRADUATE SCHOOL UNIVERSITY OF RHODE ISLAND 2014 ABSTRACT Estuaries regulate nitrogen (N) fluxes transported from land to the open ocean through uptake and denitrification. In Narragansett Bay, anthropogenic N loading has increased over the last century with evidence for eutrophication in some regions of Narragansett Bay. Increased concerns over eutrophication prompted upgrades at wastewater treatment facilities (WWTFs) to decrease the amount of nitrogen discharged. The upgrade to tertiary treatment – where bioavailable nitrogen is reduced and removed through denitrification – has occurred at multiple facilities throughout Narragansett Bay’s watershed. -

1 Introduction
1 Introduction WHO commissions reviews and undertakes health risk assessments associated with exposure to potentially hazardous physical, chemical and biological agents in the home, work place and environment. This monograph on the chemical and radiological hazards associated with exposure to depleted uranium is one such assessment. The purpose of this monograph is to provide generic information on any risks to health from depleted uranium from all avenues of exposure to the body and from any activity where human exposure could likely occur. Such activities include those involved with fabrication and use of DU products in industrial, commercial and military settings. While this monograph is primarily on DU, reference is also made to the health effects and behaviour of uranium, since uranium acts on body organs and tissues in the same way as DU and the results and conclusions from uranium studies are considered to be broadly applicable to DU. However, in the case of effects due to ionizing radiation, DU is less radioactive than uranium. This review is structured as broadly indicated in Figure 1.1, with individual chapters focussing on the identification of environmental and man-made sources of uranium and DU, exposure pathways and scenarios, likely chemical and radiological hazards and where data is available commenting on exposure-response relationships. HAZARD IDENTIFICATION PROPERTIES PHYSICAL CHEMICAL BIOLOGICAL DOSE RESPONSE RISK EVALUATION CHARACTERISATION BACKGROUND EXPOSURE LEVELS EXPOSURE ASSESSMENT Figure 1.1 Schematic diagram, depicting areas covered by this monograph. It is expected that the monograph could be used as a reference for health risk assessments in any application where DU is used and human exposure or contact could result. -

Sources of Variation in the Stable Isotopic Composition of Plants*
CHAPTER 2 Sources of variation in the stable isotopic composition of plants* JOHN D. MARSHALL, J. RENÉE BROOKS, AND KATE LAJTHA Introduction The use of stable isotopes of carbon, nitrogen, oxygen, and hydrogen to study physiological processes has increased exponentially in the past three decades. When Harmon Craig (1953, 1954), a geochemist and early pioneer of natural abundance stable isotopes, fi rst measured isotopic values of plant materials, he found that plants tended to have a fairly narrow δ13C range of −25 to −35‰. In these initial surveys, he was unable to fi nd large taxonomic or environmental effects on these values. Since that time ecologists have identi- fi ed clear isotopic signatures based not only on different photosynthetic pathways, but also on ecophysiological differences, such as photosynthetic water-use effi ciency (WUE) and sources of water and nitrogen used. As large empirical databases have accumulated and our theoretical understanding of isotopic composition has improved, scientists have continued to discover mismatches between theoretical and observed values, as well as confounding effects from sources and factors not previously considered. In the best tradi- tion of science, these discoveries have led to important new insights into physiological or ecological processes, as well as new uses of stable isotopes in plant ecophysiology. This chapter reviews the most common applications of stable isotope analysis in plant ecophysiology. Carbon isotopes Photosynthetic pathways 13 Plants contain less C than the atmospheric CO2 on which they rely for photosynthesis. They are therefore “depleted” of 13C relative to the atmo- sphere. This depletion is caused by enzymatic and physical processes that discriminate against 13C in favor of 12C. -

Characterisation of Various Types of Alloy by K0-Neutron Activation Analysis
A27 CHARACTERISATION OF VARIOUS TYPES OF ALLOY BY K0-NEUTRON ACTIVATION ANALYSIS M. WASIM, N. KHALID, M. ARIF Chemistry Division, Pakistan Institute of Nuclear Science and Technology, Islamabad, Pakistan N.A. LODHI Isotope Production Division, Pakistan Institute of Nuclear Science and Technology, Islamabad, Pakistan Abstract Samples of certified alloys were analysed by semi-absolute, standardless k0-instrumental neutron activation analysis (k0-INAA) for compositional decoding. Irradiations were performed at Miniaturised Neutron Source Reactor (MNSR) located at Pakistan Institute of Nuclear Science and Technology, Islamabad having nominal thermal neutron fluxes of 1×1012 cm-2s-1. The experimentally optimised parameters for NAA suggested a maximum of three irradiations for the quantification of 21 elements within 5 days. The same experimental conditions produced quantitative results of 13 elements, which were not reported by the supplier of the reference materials. All reference concentrations were within 95% confidence interval of the determined concentrations. 1. INTRODUCTION Worldwide interest in the determination of elements in different materials has led to the development of many analytical techniques. The commonly used techniques include inductively coupled plasma with optical emission spectrometry (ICP-OES), X-ray fluorescence spectrometry (XRF), atomic absorption spectrometry (AAS) [1], arc/spark optical emission spectrometry, ICP with mass spectrometry and laser-induced breakdown spectroscopy [2-4]. Nuclear analytical techniques also play important role in material characterisation [5]. Among these the noticeable are particle induced X-ray emission, proton activation analysis [6,7], prompt gamma-ray neutron activation [8], fast neutron activation analysis [9,10] and thermal neutron activation analysis (NAA) [11,12]. The non-nuclear techniques apply relative standardisation, also known as classical linear calibration, whereby a calibration curve is drawn by using three or more calibration standards. -

Measuring Long-Range 13C–13C Correlations on a Surface Under
Ames Laboratory Accepted Manuscripts Ames Laboratory 10-13-2017 Measuring Long-Range 13C–13C Correlations on a Surface under Natural Abundance Using Dynamic Nuclear Polarization-Enhanced Solid- State Nuclear Magnetic Resonance Takeshi Kobayashi Ames Laboratory, [email protected] Igor I. Slowing Iowa State University and Ames Laboratory, [email protected] Marek Pruski Iowa State University and Ames Laboratory, [email protected] Follow this and additional works at: http://lib.dr.iastate.edu/ameslab_manuscripts Part of the Materials Chemistry Commons, and the Physical Chemistry Commons Recommended Citation Kobayashi, Takeshi; Slowing, Igor I.; and Pruski, Marek, "Measuring Long-Range 13C–13C Correlations on a Surface under Natural Abundance Using Dynamic Nuclear Polarization-Enhanced Solid-State Nuclear Magnetic Resonance" (2017). Ames Laboratory Accepted Manuscripts. 33. http://lib.dr.iastate.edu/ameslab_manuscripts/33 This Article is brought to you for free and open access by the Ames Laboratory at Iowa State University Digital Repository. It has been accepted for inclusion in Ames Laboratory Accepted Manuscripts by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Measuring Long-Range 13C–13C Correlations on a Surface under Natural Abundance Using Dynamic Nuclear Polarization-Enhanced Solid-State Nuclear Magnetic Resonance Abstract We report that spatial (<1>nm) proximity between different molecules in solid bulk materials and, for the first time, different moieties on the surface of a catalyst, can be established without isotope enrichment by means of homonuclear CHHC solid-state nuclear magnetic resonance experiment. This 13C–13C correlation measurement, which hitherto was not possible for natural-abundance solids, was enabled by the use of dynamic nuclear polarization.