Genetic Analysis of the Hungarian Draft Horse Population Using Partial Mitochondrial DNA D-Loop Sequencing
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Multiple Choice Choose the Answer That Best Completes Each Statement Or Question
Name Date Hour 9 The Horse Industry Multiple Choice Choose the answer that best completes each statement or question. _______ 1. The horse was first domesticated in Europe and Asia about ____ . A. 1,000 years ago B. 3,000 years ago C. 5,000 years ago D. 8,000 years ago _______ 2. Horses were brought to the New World by Spanish explorers in the ____ . A. 15th century B. 16th century C. 17th century D. 18th century _______ 3. Horses are measured in terms of hands with a hand being ____ . A. two inches B. four inches C. six inches D. eight inches _______ 4. Ponies are shorter than horses and can be anywhere from 8 to ____ . A. 10 hands high B. 12.2 hands high C. 14.2 hands high D. 16 hands high _______ 5. Which horse breed is considered the oldest purebred horse in the world? A. Arabian B. Appaloosa C. Thoroughbred D. Quarter Horse Introduction to Agriscience | Unit 9 Test CIMC 1 _______ 6. Which horse breed has as one of its characteristics a distinctive spotted coat? A. Arabian B. Appaloosa C. Thoroughbred D. Quarter Horse _______ 7. Which horse breed was developed in the United States and got its name because of its great speed at short distances? A. Arabian B. Appaloosa C. Thoroughbred D. Quarter Horse _______ 8. Which horse breed was developed in the deserts of the Middle East? A. Arabian B. Appaloosa C. Thoroughbred D. Quarter Horse _______ 9. Which horse breed has a head characterized by a dished profile, prominent eye, large nostrils, and small muzzle? A. -

Analysis of Measurements of Latvian Warmblood and Latvian Heavy Warmblood Sires
AGRICULTURAL SCIENCES (CROP SCIENCES, ANIMAL SCIENCES) DOI: 10.22616/rrd.24.2018.061 ANALYSIS OF MEASUREMENTS OF LATVIAN WARMBLOOD AND LATVIAN HEAVY WARMBLOOD SIRES Anna Veidemane, Daina Jonkus Latvia University of Life Sciences and Technologies, Latvia [email protected] Abstract The objective of the study was to analyze measurements of the sires used in Latvian Warmblood (LWB) and Latvian Heavy Warmblood (LHWB) breeding programs in the period 2003 – 2017, two major horse populations in Latvia included in one studbook. The Latvian Warmblood has an open studbook for breeding sport horses, whereas the Latvian Heavy Warmblood is a partly closed studbook. Measuring information for all sires with at least one foal born (n=834) in the respective time period was retrieved from the Latvian horse database, with 673 stallions measured at least once. The data consisted of direct measurements – height at withers, chest circumference and cannon bone circumference – and two calculated indices – massivity index and boniness index. Average values of adult stallions were analyzed in four groups – LWB, LHWB, ‘other warmbloods’ and refining breeds, with LWB and ‘other warmbloods’ showing similar average values. Sires were divided by use in breeding into 3-year periods to observe a possible change in the breeding objective and stallion choice, however, no significant differences were found in LWB or LHWB. Average measurements of stallions used in the LWB breeding program (different breeds) were 168.6 ± 4.3 cm for height at withers, 194.4 ± 6.6 cm for chest circumference, 21.8 ± 1.0 cm for cannon bone circumference, massivity index 115.5 ± 3.1, boniness index 13.0 ± 0.5. -

Winter 2006 Impulsion.Indd
Winter 2006 HOLSTEINERThe Official Newsletter of the American Holsteiner Horse Association 2006 AHHA Approvals Results Home Again Fox Fire Farm We would like to thank and congratulate all breeders for showing your offspring from Fox Fire Farm stallions at the recent AHHA Approvals. Some premiums include: Photo by Paula Chamura LinaroAriadus Premium Premium Colt, Filly Lyonne Breeder: BrendanDebra Bartlik Mesker Ariadus Premium Colt, Addison Breeder: Deborah Bartlik Camiros Colt, Candelero Photo by Reg Corkum Champion, Colts & Geldings, Cool Autust Nights Breed Show, LA Equestrian Center. Ariadus Premium Filly, Uncia Reserve Best Young Horse, Hunter Breed Breeder: Andras Szieberth Show at Pebble Beach. Congratulations, KIm! Breeder: Dr. Kim Gill Favier FOX FIRE FARM 450 Fox Fire Lane NEWS: Discounted breeding fees for active PO Box 8 AHHA members, Premium mares, show winners Fox Island, WA 98333 and return breeders! E-mail: [email protected] The American Holsteiner Horse Association, Inc. 222 East Main Street, President’s Message Georgetown, KY 40324 502-863-4239, Fax: 502-868-0722 From the President: Executive Director Bruce Cottew The 2005 approvals tour is behind us. Many Registrar Jennifer Franco Executive Assistant Jennifer Walker thanks to all who showed horses and foals, Webmaster Mary Jane Gook and also all who worked behind the scenes. Special thanks go to the hard-working judg- 2005 Board of Directors President ing team of chief judge Arlene Rigdon, judges Daniel van Heeckeren 2007 440-423-3244 Eva Maria Junkelmann and Heino Kracht, and Vice President Karen Reid 2007 253-549-2838 also stallion judge Joachim Tietz at Solvang, Treasurer CA and Winley Farm, in Milbrook, NY. -
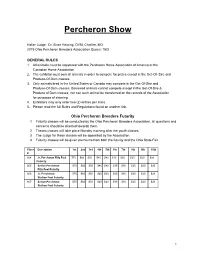
Percheron Show
Percheron Show Halter Judge: Dr. Brian Heuring, DVM, Chaffee, MO 2019 Ohio Percheron Breeders Association Queen: TBD GENERAL RULES 1. All animals must be registered with the Percheron Horse Association of America or the Canadian Horse Association 2. The exhibitor must own all animals in order to compete for prizes except in the Get-Of- Sire and Produce-Of-Dam classes. 3. Only animals bred in the United States or Canada may compete in the Get-Of-Sire and Produce-Of-Dam classes. Borrowed animals cannot compete except in the Get-Of-Sire & Produce of Dam classes, nor can such animal be transferred on the records of the Association for purposes of showing. 4. Exhibitors may only enter two (2) entries per class. 5. Please read the full Rules and Regulations found on another link. Ohio Percheron Breeders Futurity 1. Futurity classes will be conducted by the Ohio Percheron Breeders Association, all questions and concerns should be directed towards them. 2. Theses classes will take place Monday morning after the youth classes. 3. The Judge for these classes will be appointed by the Association. 4. Futurity classes will be given premiums from both the futurity and the Ohio State Fair Class Description 1st 2nd 3rd 4th 5th 6th 7th 8th 9th 10th # 604 Jr. Percheron Filly Foal $75 $60 $50 $45 $40 $35 $30 $25 $20 $20 Futurity 605 Senior Percheron $75 $60 $50 $45 $40 $35 $30 $25 $20 $20 Filly Foal Futurity 606 Jr. Percheron $75 $60 $50 $45 $40 $35 $30 $25 $20 $20 Stallion Foal Futurity 607 Senior Percheron $75 $60 $50 $45 $40 $35 $30 $25 $20 $20 Stallion Foal Futurity 1 PERCHERON HALTER CLASSES STALLIONS Entry Class Prize Description 1st 2nd 3rd 4th 5th 6th 7th 8th 9th 10th # Money Fee 625 $240 Registered Percheron Stallion, 3 Years $5 $75 $60 $50 $35 $20 & Over. -

Purebred Dog Breeds Into the Twenty-First Century: Achieving Genetic Health for Our Dogs
Purebred Dog Breeds into the Twenty-First Century: Achieving Genetic Health for Our Dogs BY JEFFREY BRAGG WHAT IS A CANINE BREED? What is a breed? To put the question more precisely, what are the necessary conditions that enable us to say with conviction, "this group of animals constitutes a distinct breed?" In the cynological world, three separate approaches combine to constitute canine breeds. Dogs are distinguished first by ancestry , all of the individuals descending from a particular founder group (and only from that group) being designated as a breed. Next they are distinguished by purpose or utility, some breeds existing for the purpose of hunting particular kinds of game,others for the performance of particular tasks in cooperation with their human masters, while yet others owe their existence simply to humankind's desire for animal companionship. Finally dogs are distinguished by typology , breed standards (whether written or unwritten) being used to describe and to recognize dogs of specific size, physical build, general appearance, shape of head, style of ears and tail, etc., which are said to be of the same breed owing to their similarity in the foregoing respects. The preceding statements are both obvious and known to all breeders and fanciers of the canine species. Nevertheless a correct and full understanding of these simple truisms is vital to the proper functioning of the entire canine fancy and to the health and well being of the animals which are the object of that fancy. It is my purpose in this brief to elucidate the interrelationship of the above three approaches, to demonstrate how distortions and misunderstandings of that interrelationship now threaten the health of all of our dogs and the very existence of the various canine breeds, and to propose reforms which will restore both balanced breed identity and genetic health to CKC breeds. -

List of Horse Breeds 1 List of Horse Breeds
List of horse breeds 1 List of horse breeds This page is a list of horse and pony breeds, and also includes terms used to describe types of horse that are not breeds but are commonly mistaken for breeds. While there is no scientifically accepted definition of the term "breed,"[1] a breed is defined generally as having distinct true-breeding characteristics over a number of generations; its members may be called "purebred". In most cases, bloodlines of horse breeds are recorded with a breed registry. However, in horses, the concept is somewhat flexible, as open stud books are created for developing horse breeds that are not yet fully true-breeding. Registries also are considered the authority as to whether a given breed is listed as Light or saddle horse breeds a "horse" or a "pony". There are also a number of "color breed", sport horse, and gaited horse registries for horses with various phenotypes or other traits, which admit any animal fitting a given set of physical characteristics, even if there is little or no evidence of the trait being a true-breeding characteristic. Other recording entities or specialty organizations may recognize horses from multiple breeds, thus, for the purposes of this article, such animals are classified as a "type" rather than a "breed". The breeds and types listed here are those that already have a Wikipedia article. For a more extensive list, see the List of all horse breeds in DAD-IS. Heavy or draft horse breeds For additional information, see horse breed, horse breeding and the individual articles listed below. -

Biodiversity of Arabian Horses in Syria
Biodiversity of Arabian horses in Syria Dissertation zur Erlangung des akademischen Grades Doctor rerum agriculturarum (Dr. rer. agr.) eingereicht an der Lebenswissenschaftlichen Fakultät der Humboldt Universität zu Berlin von M.Sc. Saria Almarzook Präsidentin der Humboldt-Universität zu Berlin Prof. Dr. Sabine Kunst Dekan der Humboldt-Universität zu Berlin Prof. Dr. Bernhard Grimm Gutachterin/Gutachter Prof. Dr. Gudrun Brockmann Prof. Dr. Dirk Hinrichs Prof. Dr. Armin Schmitt Tag der mündlichen Prüfung: 17. September 2018 Dedication This research is dedicated to my homeland …Syria Contents Zusammenfassung ................................................................................................................... I Summary ............................................................................................................................... VI List of publications and presentations .................................................................................. XII List of abbreviations ............................................................................................................ XIII List of figures ....................................................................................................................... XIV List of tables ......................................................................................................................... XV 1. General introduction and literature review ..................................................................... 1 1.1. Domestication and classification -

Special Issue Horse Genetics DIRECTOR’S Message
CENTER FOR EQUINE HEALTH SCHOOL OF VETERINARY MEDICINE • UNIVERSITY OF CALIFORNIA, DAVIS SUMMER 2020 Special Issue Horse Genetics DIRECTOR’S Message s an equine genetics researcher, I am particularly excited to share A this special issue of the Horse Report with you. Inside, you will find a roadmap to many of the currently available equine genetic tests, including the AQHA “five-panel” test, and more. The equine genome sequence was published in 2009, the result of a years- long collaborative effort by the international equine research community. This resource drastically changed how researchers approach equine genetics and accelerated the rate of discovery. Increased availability and affordability allowed the application of advanced molecular tools to equine diseases and traits. As a result, genetic tests are available in a variety of breeds. Most available tests are for simple, Mendelian diseases and traits – those caused by a single gene or locus. Complex diseases and traits likely involve more than one gene and may be influenced by environmental effects. The 2018 release of a new equine genome sequence assembly, coupled with cost reductions that make whole-genome sequencing possible for large numbers of horses, are enabling research in these areas. As an equine geneticist and veterinarian, I am especially interested in applying whole genome sequencing and advanced diagnostic tools to equine precision medicine. This highly individualized approach will focus on early detection and prevention of disease, taking into account both genetic information and environmental factors. The idea is to target individuals based on their clinical condition as well as their unique body chemistry and genetics. -

A View of the Horse from the Classical Perspective the Penn Museum Collection by Donald White
A View of the Horse from the Classical Perspective The Penn Museum Collection by donald white quus caballus is handsomely stabled in tive how-to manual On Horsemanship (“The tail and mane the University of Pennsylvania Museum of should be washed, to keep the hairs growing, as the tail is used Archaeology and Anthropology. From the to swat insects and the mane may be grabbed by the rider Chinese Rotunda’s masterpiece reliefs portray- more easily if long.”) all the way down to the 9th century AD ing two horses of the Chinese emperor Taizong Corpus of Greek Horse Veterinarians, which itemizes drugs for Eto Edward S. Curtis’s iconic American Indian photographs curing equine ailments as well as listing vets by name, Greek housed in the Museum’s Archives, horses stand with man and Roman literature is filled with equine references. One in nearly every culture and time-frame represented in the recalls the cynical utterance of the 5th century BC lyric poet Museum’s Collection (pre-Columbian America and the Xenophanes from the Asia Minor city of Colophon: “But if northern polar region being perhaps the two most obvious cattle and horses and lions had hands, or were able to do the exceptions). Examples drawn from the more than 30,000 work that men can, horses would draw the forms of the gods like Greek, Roman, and Etruscan vases, sculptures, and other horses” (emphasis added by author). objects in the Museum’s Mediterranean Section serve here as The partnership between horse and master in antiquity rested a lens through which to view some of the notable roles the on many factors; perhaps the most important was that the horse horse played in the classical Mediterranean world. -
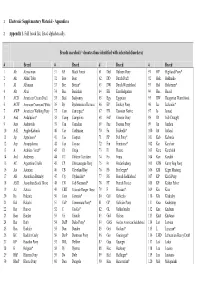
Electronic Supplementary Material - Appendices
1 Electronic Supplementary Material - Appendices 2 Appendix 1. Full breed list, listed alphabetically. Breeds searched (* denotes those identified with inherited disorders) # Breed # Breed # Breed # Breed 1 Ab Abyssinian 31 BF Black Forest 61 Dul Dülmen Pony 91 HP Highland Pony* 2 Ak Akhal Teke 32 Boe Boer 62 DD Dutch Draft 92 Hok Hokkaido 3 Al Albanian 33 Bre Breton* 63 DW Dutch Warmblood 93 Hol Holsteiner* 4 Alt Altai 34 Buc Buckskin 64 EB East Bulgarian 94 Huc Hucul 5 ACD American Cream Draft 35 Bud Budyonny 65 Egy Egyptian 95 HW Hungarian Warmblood 6 ACW American Creme and White 36 By Byelorussian Harness 66 EP Eriskay Pony 96 Ice Icelandic* 7 AWP American Walking Pony 37 Cam Camargue* 67 EN Estonian Native 97 Io Iomud 8 And Andalusian* 38 Camp Campolina 68 ExP Exmoor Pony 98 ID Irish Draught 9 Anv Andravida 39 Can Canadian 69 Fae Faeroes Pony 99 Jin Jinzhou 10 A-K Anglo-Kabarda 40 Car Carthusian 70 Fa Falabella* 100 Jut Jutland 11 Ap Appaloosa* 41 Cas Caspian 71 FP Fell Pony* 101 Kab Kabarda 12 Arp Araappaloosa 42 Cay Cayuse 72 Fin Finnhorse* 102 Kar Karabair 13 A Arabian / Arab* 43 Ch Cheju 73 Fl Fleuve 103 Kara Karabakh 14 Ard Ardennes 44 CC Chilean Corralero 74 Fo Fouta 104 Kaz Kazakh 15 AC Argentine Criollo 45 CP Chincoteague Pony 75 Fr Frederiksborg 105 KPB Kerry Bog Pony 16 Ast Asturian 46 CB Cleveland Bay 76 Fb Freiberger* 106 KM Kiger Mustang 17 AB Australian Brumby 47 Cly Clydesdale* 77 FS French Saddlebred 107 KP Kirdi Pony 18 ASH Australian Stock Horse 48 CN Cob Normand* 78 FT French Trotter 108 KF Kisber Felver 19 Az Azteca -

Morgan Horses
The 12th Annual NATIONAL MORGAN HORSE SHOW Sponsored by: Saturday Evening Friday Evening 7:00 P. M. 7:00 P. M. Sunday Saturday Afternoon Afternoon 1:00 P. M. 1:00 P. M. PERFORMANCE BREED CLASSES CLASSES For Stallions and Saddle, Harness, Mares: Colts and Pleasure. Utility Fillies and Equitation THE MORGAN HORSE CLUB Watch The Foundation Breed of America Perform. TRI-COUNTY FAIR GROUNDS NORTHAMPTON, MASS. July 30, 31 and August 1, 1954 Adults $1.00 Children - under 12 - 50' A LAW FOR IT . by 1939 Vermont Legislature "There oughta be a law agin it," is a favorite expresion of Vermonters. Sometimes they reverse themselves and make a law "for it" as they did in 1939 when the legislature passed the following resolution: "Whereas, this is the year recognized as the 150th anniversa y of the famous horse 'Justin Morgan,' which horse not only established a recognized breed of horses named for a single individual, but brought fame th•tzugh his descendants to Vermont and thousands of dollars to Vermonters. "The name Morgan has come to mean beauty, spirit, and action to all lovers of the horse; and the Morgan horses fo• many years held the world's record for trotting horses, and "Whereas the Morgan blood is recognized as foundation stock for the American Saddle Horse, for the American Trotting Horse, and for the Tennessee Walking Horse. In each of these three breeds, the Morgan horse is recognized as a foundation, and therefore, with the recognition of its value to the horse b seeders of the nation, and recognition that it was in Vermont that Morgan -

Draft Horse Handbook
EB1135E Draft Horse Handbook WASHINGTON STATE UNIVERSITY EXTENSION CONTENTS Breeds of Draft Horses ................................................................................................. 1 Belgian ...................................................................................................................... 1 Percheron .................................................................................................................. 1 Clydesdale ................................................................................................................. 2 Shire .......................................................................................................................... 3 Suffolk ....................................................................................................................... 3 Mule .......................................................................................................................... 4 Draft Horse Judging ..................................................................................................... 4 Showing Draft Horses at Halter .................................................................................. 7 The Handler ............................................................................................................... 7 The Horse .................................................................................................................. 7 In the Ring ................................................................................................................