Fatty Acid Synthase: an Emerging Target in Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Amphibolic Nature of Krebs Cycle
Amphibolic nature of Krebs Cycle How what we are is what we eat • In aerobic organisms, the citric acid cycle is an amphibolic pathway, one that serves in both catabolic and anabolic processes. • Since the citric acid does both synthesis (anabolic) and breakdown (catabolic) activities, it is called an amphibolic pathway • The citric acid cycle is amphibolic (i.e it is both anabolic and catabolic in its function). • It is said to be an AMPHIBOLIC pathway, because it functions in both degradative or catabolic and biosynthetic or anabolic reactions (amphi = both) A central metabolic pathway or amphibolic pathway is a set of reactions which permit the interconversion of several metabolites, and represents the end of the catabolism and the beginning of anabolism • The KREBS CYCLE or citric acid cycle is a series of reactions that degrades acetyl CoA to yield carbon dioxide, and energy, which is used to produce NADH, H+ and FADH. • The KREBS CYCLE connects the catabolic pathways that begin with the digestion and degradation of foods in stages 1 and 2 with the oxidation of substrates in stage 3 that generates most of the energy for ATP synthesis. • The citric acid cycle is the final common pathway in the oxidation of fuel molecules. In stage 3 of metabolism, citric acid is a final common catabolic intermediate in the form of acetylCoA. • This is why the citric acid cycle is called a central metabolic pathway. Anaplerosis and Cataplerosis Anaplerosis is a series of enzymatic reactions in which metabolic intermediates enter the citric acid cycle from the cytosol. Cataplerosis is the opposite, a process where intermediates leave the citric acid cycle and enter the cytosol. -

Fructose Metabolism from a Functional
SSE #174 Sports Science Exchange (2017) Vol. 28, No. 174, 1-5 FRUCTOSE METABOLISM FROM A FUNCTIONAL PERSPECTIVE: IMPLICATIONS FOR ATHLETES Luke Tappy, MD | Department of Physiology, Faculty of Biology and Medicine, University of Lausanne, Service of Endocrinology, Diabetes and Metabolism | Lausanne University Hospital, and Cardio-metabolic Center, Broye Hospital | Estavayer-le-lac, Switzerland • Fructose was originally a seasonal natural nutrient, mainly consumed in summer and fall in fruits and vegetables. In the industrial era, it became a permanent constituent of our diet, essentially a constituent of added sugars (sucrose, high-fructose corn syrup). • Fructose cannot be directly metabolized by most cells in our body. It has to be processed first in the gut, liver and kidneys, where it is converted into glucose, lactate and fatty acids. • Too much dietary fructose along with excess energy intake and low physical activity can cause hepatic insulin resistance, hypertriglyceridemia and increased hepatic fat content. GAT11LOGO_GSSI_vert_fc_grn • In exercising athletes, net carbohydrate oxidation increases with glucose ingestion in a dose-dependent manner until a plateau is reached at about 1g/min. The addition of fructose to glucose drinks can further increase carbohydrate oxidation. • During exercise, substantial amounts of fructose can be converted into lactate in splanchnic organs if available and released in the systemic circulation to be oxidized in contracting muscles. This “reverse fructose-lactate Cori cycle” provides additional energy substrate to muscle during exercise. • Conversion of fructose into glucose and lactate in splanchnic organs is associated with enhanced splanchnic energy expenditure, while muscle energy efficiency is minimally altered. • During recovery after exercise, glucose and fructose mutually enhance their gut absorption and their storage as glycogen in the liver. -

• Glycolysis • Gluconeogenesis • Glycogen Synthesis
Carbohydrate Metabolism! Wichit Suthammarak – Department of Biochemistry, Faculty of Medicine Siriraj Hospital – Aug 1st and 4th, 2014! • Glycolysis • Gluconeogenesis • Glycogen synthesis • Glycogenolysis • Pentose phosphate pathway • Metabolism of other hexoses Carbohydrate Digestion! Digestive enzymes! Polysaccharides/complex carbohydrates Salivary glands Amylase Pancreas Oligosaccharides/dextrins Dextrinase Membrane-bound Microvilli Brush border Maltose Sucrose Lactose Maltase Sucrase Lactase ‘Disaccharidase’ 2 glucose 1 glucose 1 glucose 1 fructose 1 galactose Lactose Intolerance! Cause & Pathophysiology! Normal lactose digestion Lactose intolerance Lactose Lactose Lactose Glucose Small Intestine Lactase lactase X Galactose Bacteria 1 glucose Large Fermentation 1 galactose Intestine gases, organic acid, Normal stools osmotically Lactase deficiency! active molecules • Primary lactase deficiency: อาการ! genetic defect, การสราง lactase ลด ลงเมออายมากขน, พบมากทสด! ปวดทอง, ถายเหลว, คลนไสอาเจยนภาย • Secondary lactase deficiency: หลงจากรบประทานอาหารทม lactose acquired/transient เชน small bowel เปนปรมาณมาก เชนนม! injury, gastroenteritis, inflammatory bowel disease! Absorption of Hexoses! Site: duodenum! Intestinal lumen Enterocytes Membrane Transporter! Blood SGLT1: sodium-glucose transporter Na+" Na+" •! Presents at the apical membrane ! of enterocytes! SGLT1 Glucose" Glucose" •! Co-transports Na+ and glucose/! Galactose" Galactose" galactose! GLUT2 Fructose" Fructose" GLUT5 GLUT5 •! Transports fructose from the ! intestinal lumen into enterocytes! -

Altered Expression and Function of Mitochondrial Я-Oxidation Enzymes
0031-3998/01/5001-0083 PEDIATRIC RESEARCH Vol. 50, No. 1, 2001 Copyright © 2001 International Pediatric Research Foundation, Inc. Printed in U.S.A. Altered Expression and Function of Mitochondrial -Oxidation Enzymes in Juvenile Intrauterine-Growth-Retarded Rat Skeletal Muscle ROBERT H. LANE, DAVID E. KELLEY, VLADIMIR H. RITOV, ANNA E. TSIRKA, AND ELISA M. GRUETZMACHER Department of Pediatrics, UCLA School of Medicine, Mattel Children’s Hospital at UCLA, Los Angeles, California 90095, U.S.A. [R.H.L.]; and Departments of Internal Medicine [D.E.K., V.H.R.] and Pediatrics [R.H.L., A.E.T., E.M.G.], University of Pittsburgh School of Medicine, Magee-Womens Research Institute, Pittsburgh, Pennsylvania 15213, U.S.A. ABSTRACT Uteroplacental insufficiency and subsequent intrauterine creased in IUGR skeletal muscle mitochondria, and isocitrate growth retardation (IUGR) affects postnatal metabolism. In ju- dehydrogenase activity was unchanged. Interestingly, skeletal venile rats, IUGR alters skeletal muscle mitochondrial gene muscle triglycerides were significantly increased in IUGR skel- expression and reduces mitochondrial NADϩ/NADH ratios, both etal muscle. We conclude that uteroplacental insufficiency alters of which affect -oxidation flux. We therefore hypothesized that IUGR skeletal muscle mitochondrial lipid metabolism, and we gene expression and function of mitochondrial -oxidation en- speculate that the changes observed in this study play a role in zymes would be altered in juvenile IUGR skeletal muscle. To test the long-term morbidity associated with IUGR. (Pediatr Res 50: this hypothesis, mRNA levels of five key mitochondrial enzymes 83–90, 2001) (carnitine palmitoyltransferase I, trifunctional protein of -oxi- dation, uncoupling protein-3, isocitrate dehydrogenase, and mi- Abbreviations tochondrial malate dehydrogenase) and intramuscular triglycer- CPTI, carnitine palmitoyltransferase I ides were quantified in 21-d-old (preweaning) IUGR and control IUGR, intrauterine growth retardation rat skeletal muscle. -

ATP-Citrate Lyase Has an Essential Role in Cytosolic Acetyl-Coa Production in Arabidopsis Beth Leann Fatland Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 2002 ATP-citrate lyase has an essential role in cytosolic acetyl-CoA production in Arabidopsis Beth LeAnn Fatland Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Molecular Biology Commons, and the Plant Sciences Commons Recommended Citation Fatland, Beth LeAnn, "ATP-citrate lyase has an essential role in cytosolic acetyl-CoA production in Arabidopsis " (2002). Retrospective Theses and Dissertations. 1218. https://lib.dr.iastate.edu/rtd/1218 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. ATP-citrate lyase has an essential role in cytosolic acetyl-CoA production in Arabidopsis by Beth LeAnn Fatland A dissertation submitted to the graduate faculty in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY Major: Plant Physiology Program of Study Committee: Eve Syrkin Wurtele (Major Professor) James Colbert Harry Homer Basil Nikolau Martin Spalding Iowa State University Ames, Iowa 2002 UMI Number: 3158393 INFORMATION TO USERS The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleed-through, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. -

Multiple N-Acetyltransferases and Drug Metabolism TISSUE DISTRIBUTION, CHARACTERIZATION and SIGNIFICANCE of MAMMALIAN N-ACETYLTRANSFERASE by D
Biochem. J. (1973) 132, 519-526 519 Printed in Great Britain Multiple N-Acetyltransferases and Drug Metabolism TISSUE DISTRIBUTION, CHARACTERIZATION AND SIGNIFICANCE OF MAMMALIAN N-ACETYLTRANSFERASE By D. J. HEARSE* and W. W. WEBER Department ofPharmacology, New York University Medical Center, 550 First Avenue, New York, N. Y. 10016, U.S.A. (Received 6 October 1972) Investigations in the rabbit have indicated the existence of more than one N-acetyl- transferase (EC 2.3.1.5). At least two enzymes, possibly isoenzymes, were partially characterized. The enzymes differed in their tissue distribution, substrate specificity, stability and pH characteristics. One of the enzymes was primarily associated with liver and gut and catalysed the acetylation of a wide range of drugs and foreign compounds, e.g. isoniazid, p-aminobenzoic acid, sulphamethazine and sulphadiazine. The activity of this enzyme corresponded to the well-characterized polymorphic trait of isoniazid acetylation, and determined whether individuals were classified as either 'rapid' or 'slow' acetylators. Another enzyme activity found in extrahepatic tissues readily catalysed the acetylation ofp-aminobenzoic acid but was much less active towards isoniazid and sulpha- methazine. The activity of this enzyme remained relatively constant from individual to individual. Studies in vitro and in vivo with both 'rapid' and 'slow' acetylator rabbits re- vealed that, for certain substrates, extrahepatic N-acetyltransferase contributes signifi- cantly to the total acetylating capacity of the individual. The possible significance and applicability ofthese findings to drugmetabolism and acetylation polymorphism in man is discussed. Liver N-acetyltransferase catalyses the acetylation purified by the same procedure and their pH charac- of a number of commonly used drugs and foreign teristics, heat stabilities, kinetic properties, substrate compounds such as isoniazid, sulphamethazine, specificities and reaction mechanisms are indis- sulphadiazine, p-aminobenzoic acid, diamino- tinguishable. -

Process for Production of Lipstatin and Microorganisms Therefore
(19) *EP002141236A1* (11) EP 2 141 236 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 06.01.2010 Bulletin 2010/01 C12N 15/52 (2006.01) A61K 31/365 (2006.01) C12P 17/02 (2006.01) C07D 305/12 (2006.01) (21) Application number: 08012016.5 (22) Date of filing: 03.07.2008 (84) Designated Contracting States: • Kuscer, Enej AT BE BG CH CY CZ DE DK EE ES FI FR GB GR 1000 Ljubljana (SI) HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT • Petrovic, Hrvoje RO SE SI SK TR 1000 Ljubljana (SI) Designated Extension States: • Sladic, Gordan AL BA MK RS 8000 Novo mesto (SI) • Gasparic, Ales (83) Declaration under Rule 32(1) EPC (expert 8000 Novo mesto (SI) solution) (74) Representative: HOFFMANN EITLE (71) Applicant: KRKA, D.D., Novo Mesto Patent- und Rechtsanwälte 8501 Novo mesto (SI) Arabellastrasse 4 81925 München (DE) (72) Inventors: • Fujs, Stefan 9201 Puconci (SI) (54) Process for production of lipstatin and microorganisms therefore (57) This invention relates to improved processes for having reduced or abolished BKD activity. The invention the production of lipstatin and /or orlistat and means in particular relates to lipstatin-producing microorgan- therefore. In particular, the invention relates to microor- isms of the genus Streptomyces. Furthermore, this in- ganisms characterized by abolished or reduced activity vention relates to the use of these nucleotide sequences, of the BKD complex, e.g. mutated branched-chain 2-oxo microorganisms or BKD having reduced or abolished ac- acid dehydrogenase (BKD) having reduced or abolished tivity in the production of lipstatin and/or orlistat. -

Contig Protein Description Symbol Anterior Posterior Ratio
Table S2. List of proteins detected in anterior and posterior intestine pooled samples. Data on protein expression are mean ± SEM of 4 pools fed the experimental diets. The number of the contig in the Sea Bream Database (http://nutrigroup-iats.org/seabreamdb) is indicated. Contig Protein Description Symbol Anterior Posterior Ratio Ant/Pos C2_6629 1,4-alpha-glucan-branching enzyme GBE1 0.88±0.1 0.91±0.03 0.98 C2_4764 116 kDa U5 small nuclear ribonucleoprotein component EFTUD2 0.74±0.09 0.71±0.05 1.03 C2_299 14-3-3 protein beta/alpha-1 YWHAB 1.45±0.23 2.18±0.09 0.67 C2_268 14-3-3 protein epsilon YWHAE 1.28±0.2 2.01±0.13 0.63 C2_2474 14-3-3 protein gamma-1 YWHAG 1.8±0.41 2.72±0.09 0.66 C2_1017 14-3-3 protein zeta YWHAZ 1.33±0.14 4.41±0.38 0.30 C2_34474 14-3-3-like protein 2 YWHAQ 1.3±0.11 1.85±0.13 0.70 C2_4902 17-beta-hydroxysteroid dehydrogenase 14 HSD17B14 0.93±0.05 2.33±0.09 0.40 C2_3100 1-acylglycerol-3-phosphate O-acyltransferase ABHD5 ABHD5 0.85±0.07 0.78±0.13 1.10 C2_15440 1-phosphatidylinositol phosphodiesterase PLCD1 0.65±0.12 0.4±0.06 1.65 C2_12986 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase delta-1 PLCD1 0.76±0.08 1.15±0.16 0.66 C2_4412 1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase gamma-2 PLCG2 1.13±0.08 2.08±0.27 0.54 C2_3170 2,4-dienoyl-CoA reductase, mitochondrial DECR1 1.16±0.1 0.83±0.03 1.39 C2_1520 26S protease regulatory subunit 10B PSMC6 1.37±0.21 1.43±0.04 0.96 C2_4264 26S protease regulatory subunit 4 PSMC1 1.2±0.2 1.78±0.08 0.68 C2_1666 26S protease regulatory subunit 6A PSMC3 1.44±0.24 1.61±0.08 -

Short Chain Fatty Acid Biosynthesis in Microalgae Synechococcus Sp. PCC 7942
marine drugs Article Short Chain Fatty Acid Biosynthesis in Microalgae Synechococcus sp. PCC 7942 Yi Gong 1,2,3 and Xiaoling Miao 1,2,3,* 1 State Key Laboratory of Microbial Metabolism, School of Life Sciences & Biotechnology, Shanghai Jiao Tong University, 800 Dongchuan Road, Shanghai 200240, China; [email protected] 2 Joint International Research Laboratory of Metabolic & Developmental Sciences, Shanghai Jiao Tong University, Shanghai 200240, China 3 Biomass Energy Research Center, Shanghai Jiao Tong University, Shanghai 200240, China * Correspondence: [email protected]; Tel.: +86-21-34207028 Received: 19 April 2019; Accepted: 25 April 2019; Published: 28 April 2019 Abstract: Short chain fatty acids (SCFAs) are valued as a functional material in cosmetics. Cyanobacteria can accumulate SCFAs under some conditions, the related mechanism is unclear. Two potential genes Synpcc7942_0537 (fabB/F) and Synpcc7942_1455 (fabH) in Synechococcus sp. PCC 7942 have homology with fabB/F and fabH encoding β-ketoacyl ACP synthases (I/II/III) in plants. Therefore, effects of culture time and cerulenin on SCFAs accumulation, expression levels and functions of these two potential genes were studied. The results showed Synechococcus sp. PCC 7942 accumulated high SCFAs (C12 + C14) in early growth stage (day 4) and at 7.5g/L cerulenin concentration, reaching to 2.44% and 2.84% of the total fatty acids respectively, where fabB/F expression was down-regulated. Fatty acid composition analysis showed C14 increased by 65.19% and 130% respectively, when fabB/F and fabH were antisense expressed. C14 increased by 10.79% (fab(B/F)−) and 6.47% (fabH−) under mutation conditions, while C8 increased by six times in fab(B/F)− mutant strain. -

Supplementary Materials
1 Supplementary Materials: Supplemental Figure 1. Gene expression profiles of kidneys in the Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice. (A) A heat map of microarray data show the genes that significantly changed up to 2 fold compared between Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice (N=4 mice per group; p<0.05). Data show in log2 (sample/wild-type). 2 Supplemental Figure 2. Sting signaling is essential for immuno-phenotypes of the Fcgr2b-/-lupus mice. (A-C) Flow cytometry analysis of splenocytes isolated from wild-type, Fcgr2b-/- and Fcgr2b-/-. Stinggt/gt mice at the age of 6-7 months (N= 13-14 per group). Data shown in the percentage of (A) CD4+ ICOS+ cells, (B) B220+ I-Ab+ cells and (C) CD138+ cells. Data show as mean ± SEM (*p < 0.05, **p<0.01 and ***p<0.001). 3 Supplemental Figure 3. Phenotypes of Sting activated dendritic cells. (A) Representative of western blot analysis from immunoprecipitation with Sting of Fcgr2b-/- mice (N= 4). The band was shown in STING protein of activated BMDC with DMXAA at 0, 3 and 6 hr. and phosphorylation of STING at Ser357. (B) Mass spectra of phosphorylation of STING at Ser357 of activated BMDC from Fcgr2b-/- mice after stimulated with DMXAA for 3 hour and followed by immunoprecipitation with STING. (C) Sting-activated BMDC were co-cultured with LYN inhibitor PP2 and analyzed by flow cytometry, which showed the mean fluorescence intensity (MFI) of IAb expressing DC (N = 3 mice per group). 4 Supplemental Table 1. Lists of up and down of regulated proteins Accession No. -

Inhibition of the Fungal Fatty Acid Synthase Type I Multienzyme Complex
Inhibition of the fungal fatty acid synthase type I multienzyme complex Patrik Johansson*, Birgit Wiltschi*, Preeti Kumari†, Brigitte Kessler*, Clemens Vonrhein‡, Janet Vonck†, Dieter Oesterhelt*§, and Martin Grininger*§ *Department of Membrane Biochemistry, Max Planck Institute of Biochemistry, Am Klopferspitz 18, 82152 Martinsried, Germany; †Department of Structural Biology, Max Planck Institute of Biophysics, Max-von-Laue Strasse 3, 60438 Frankfurt, Germany; and ‡Global Phasing Ltd., Sheraton House, Castle Park, Cambridge CB3 0AX, United Kingdom Communicated by Hartmut Michel, Max Planck Institute for Biophysics, Frankfurt, Germany, June 23, 2008 (received for review March 6, 2008) Fatty acids are among the major building blocks of living cells, isoniazid and triclosan, both inhibiting the ER step of bacterial making lipid biosynthesis a potent target for compounds with fatty acid biosynthesis (6, 7). Several inhibitors targeting the antibiotic or antineoplastic properties. We present the crystal ketoacyl synthase (KS) step of the FAS cycle have also been structure of the 2.6-MDa Saccharomyces cerevisiae fatty acid syn- described, including cerulenin (CER) (8), thiolactomycin (TLM) thase (FAS) multienzyme in complex with the antibiotic cerulenin, (9), and the recently discovered platensimycin (PLM) (10). The representing, to our knowledge, the first structure of an inhibited polyketide CER inhibits both FAS type I and II KS enzymes, by fatty acid megasynthase. Cerulenin attacks the FAS ketoacyl syn- covalent modification of the active site cysteine and by occupying thase (KS) domain, forming a covalent bond to the active site the long acyl-binding pocket (11, 12). TLM and PLM, in contrast, cysteine C1305. The inhibitor binding causes two significant con- have been shown to be selective toward the FAS II system, formational changes of the enzyme. -
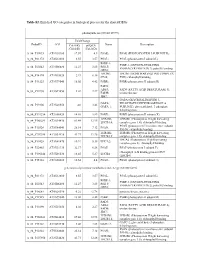
Table S2. Enriched GO Categories in Biological Process for the Shared Degs
Table S2. Enriched GO categories in biological process for the shared DEGs photosynthesis (GO ID:15979) Fold Change ProbeID AGI Col-0(R) pifQ(D) Name Description /Col-0(D) /Col-0(D) A_84_P19035 AT1G30380 17.07 4.9 PSAK; PSAK (PHOTOSYSTEM I SUBUNIT K) A_84_P21372 AT4G12800 8.55 3.57 PSAL; PSAL (photosystem I subunit L) PSBP-1; PSBP-1 (OXYGEN-EVOLVING A_84_P20343 AT1G06680 12.27 3.85 PSII-P; ENHANCER PROTEIN 2); poly(U) binding OEE2; LHCB6; LHCB6 (LIGHT HARVESTING COMPLEX A_84_P14174 AT1G15820 23.9 6.16 CP24; PSII); chlorophyll binding A_84_P11525 AT1G79040 16.02 4.42 PSBR; PSBR (photosystem II subunit R) FAD5; ADS3; FAD5 (FATTY ACID DESATURASE 5); A_84_P19290 AT3G15850 4.02 2.27 FADB; oxidoreductase JB67; GAPA (GLYCERALDEHYDE 3- GAPA; PHOSPHATE DEHYDROGENASE A A_84_P19306 AT3G26650 4.6 3.43 GAPA-1; SUBUNIT); glyceraldehyde-3-phosphate dehydrogenase A_84_P193234 AT2G06520 14.01 3.89 PSBX; PSBX (photosystem II subunit X) LHB1B1; LHB1B1 (Photosystem II light harvesting A_84_P160283 AT2G34430 89.44 32.95 LHCB1.4; complex gene 1.4); chlorophyll binding PSAN (photosystem I reaction center subunit A_84_P10324 AT5G64040 26.14 7.12 PSAN; PSI-N); calmodulin binding LHB1B2; LHB1B2 (Photosystem II light harvesting A_84_P207958 AT2G34420 41.71 12.26 LHCB1.5; complex gene 1.5); chlorophyll binding LHCA2 (Photosystem I light harvesting A_84_P19428 AT3G61470 10.91 5.36 LHCA2; complex gene 2); chlorophyll binding A_84_P22465 AT1G31330 32.37 6.58 PSAF; PSAF (photosystem I subunit F) chlorophyll A-B binding protein CP29 A_84_P190244 AT5G01530 16.45 5.27 LHCB4