TRADD–TRAF2 and TRADD–FADD Interactions Define Two Distinct TNF Receptor 1 Signal Transduction Pathways
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Accumulation of P53 and Reductions in XIAP Abundance Promote the Apoptosis of Prostate Cancer Cells
Research Article Accumulation of p53 and Reductions in XIAP Abundance Promote the Apoptosis of Prostate Cancer Cells Subhra Mohapatra, Baoky Chu, Xiuhua Zhao, and W.J. Pledger Department of Interdisciplinary Oncology, H. Lee Moffitt Cancer Center and Research Institute and the University of South Florida Medical Center, Tampa, Florida Abstract activates caspase-9. Most drugs signal apoptosis through the Toward the goal of developing effective treatments for mitochondrial pathway. prostate cancers, we examined the effects of cyclin-dependent Proteins that modulate caspase activity and thus determine kinase inhibitors on the survival of prostate cancer cells. We whether cells live or die include the inhibitor of apoptosis proteins show that roscovitine, R-roscovitine, and CGP74514A (collec- (IAPs) and the Bcl-2 proteins. The IAP family includes cIAP-1, cIAP- tively referred to as CKIs) induce the apoptosis of LNCaP and 2, XIAP, and survivin (11). Of these proteins, XIAP is the most potent. LNCaP-Rf cells, both of which express wild-type p53. Apoptosis IAPs interact with and inhibit the activity of processed caspases; required caspase-9 and caspase-3 activity, and cytochrome c thus, they function as ‘‘brakes’’ that can impede the apoptotic accumulated in the cytosol of CKI-treated cells. Amounts of process once it begins. IAPs inactivate both initiator and effector caspases; caspase-9 and caspase-3 are IAP targets, whereas caspase- p53 increased substantially in CKI-treated cells, whereas amounts of the endogenous caspase inhibitor XIAP decreased. 8 is not (12). CKIs did not appreciably induce the apoptosis of LNCaP cells The Bcl-2 proteins are critical determinants of mitochondria- treated with pifithrin-A, which prevents p53 accumulation, or dependent caspase activation (13). -

A TRAF2 Binding Independent Region of TNFR2 Is Responsible for TRAF2 Depletion and Enhancement of Cytotoxicity Driven by TNFR1
www.impactjournals.com/oncotarget/ Oncotarget, January, Vol. 5, No. 1 A TRAF2 binding independent region of TNFR2 is responsible for TRAF2 depletion and enhancement of cytotoxicity driven by TNFR1 Lucía Cabal-Hierro1,3, Noelia Artime1, Julián Iglesias1, Miguel A. Prado1,4, Lorea Ugarte-Gil1, Pedro Casado1,5, Belén Fernández-García1, Bryant G. Darnay2 and Pedro S. Lazo1. 1 Departamento de Bioquímica y Biología Molecular and Instituto Universitario de Oncología del Principado de Asturias (IUOPA), Universidad de Oviedo, Oviedo, Spain. 2 University of Texas. MD Anderson Cancer Center. Houston, Texas, USA. 3 Present address: Abramson Cancer Center. Perelman School of Medicine, University of Pennsylvania Philadelphia, PA. USA. 4 Present address: Department of Cell Biology. Harvard Medical School. Boston, MA. USA. 5 Present address: Institute of Cancer, Barts Cancer Institute. Queen Mary University of London. London, UK Correspondence to: Pedro S. Lazo, email:[email protected] Keywords: TNF receptors; Death Receptors; TRAF2; Apoptosis; NF-kappaB Received: October 11, 2013 Accepted: November 27, 2013 Published: November 29, 2013 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT: Tumor Necrosis Factor (TNF) interacts with two receptors known as TNFR1 and TNFR2. TNFR1 activation may result in either cell proliferation or cell death. TNFR2 activates Nuclear Factor-kappaB (NF-kB) and c-Jun N-terminal kinase (JNK) which lead to transcriptional activation of genes related to cell proliferation and survival. This depends on the binding of TNF Receptor Associated Factor 2 (TRAF2) to the receptor. -

Life, Death, and the Pursuit of Apoptosis
Downloaded from genesdev.cshlp.org on October 4, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Life, death, and the pursuit of apoptosis Eileen White Center for Advanced Biotechnology and Medicine and Department of Biological Sciences and the Cancer Institute of New Jersey, Rutgers University, Piscataway, New Jersey 08854 USA Apoptosis or programmed cell death is a genetically con- (Oltvai et al. 1993). Bcl-2 is expressed widely during em- trolled response for cells to commit suicide. The symp- bryogenesis (LeBrun et al. 1993; Novack and Korsmeyer toms of apoptosis are viability loss accompanied by cy- 1994), and its expression becomes more restricted in toplasmic boiling, chromatin condensation, and DNA adult tissues to those requiring long-term survival (stem fragmentation (Wyllie 1980). Pathologists and develop- cells, postmitotic neurons, and proliferating zones; mental biologists have cataloged the occurrences of ap- Hockenbery et al. 1991). optosis for many years based on these defined morpho- Apoptosis plays a major role in normal development, logical features, but what has propelled apoptosis into and it was expected that gain- or loss-of-function of a the forefront of basic research has been the identification death inhibitor such as Bcl-2 would have a phenotype in of genes that control cell death and the appreciation of transgenic mice. Bcl-2 was expected to regulate apopto- the role of apoptosis in development and disease. Regu- sis in lymphoid cells because of the role of Bcl-2 in hu- lation of cell death is essential for normal development man B cell lymphoma. Targeted Bcl-2 overexpression to and is an important defense against viral infection and the lymphoid system extends normal B cell survival the emergence of cancer. -

Combination of the Natural Compound Periplocin and TRAIL Induce
Han et al. Journal of Experimental & Clinical Cancer Research (2019) 38:501 https://doi.org/10.1186/s13046-019-1498-z RESEARCH Open Access Combination of the natural compound Periplocin and TRAIL induce esophageal squamous cell carcinoma apoptosis in vitro and in vivo: Implication in anticancer therapy Lujuan Han1†, Suli Dai1†, Zhirong Li1, Cong Zhang1, Sisi Wei1, Ruinian Zhao1, Hongtao Zhang2, Lianmei Zhao1* and Baoen Shan1* Abstract Background: Esophageal cancer is one of the most common malignant tumors in the world. With currently available therapies, only 20% ~ 30% patients can survive this disease for more than 5 years. TRAIL, a natural ligand for death receptors that can induce the apoptosis of cancer cells, has been explored as a therapeutic agent for cancers, but it has been reported that many cancer cells are resistant to TRAIL, limiting the potential clinical use of TRAIL as a cancer therapy. Meanwhile, Periplocin (CPP), a natural compound from dry root of Periploca sepium Bge, has been studied for its anti-cancer activity in a variety of cancers. It is not clear whether CPP and TRAIL can have activity on esophageal squamous cell carcinoma (ESCC) cells, or whether the combination of these two agents can have synergistic activity. Methods: We used MTS assay, flow cytometry and TUNEL assay to detect the effects of CPP alone or in combination with TRAIL on ESCC cells. The mechanism of CPP enhances the activity of TRAIL was analyzed by western blot, dual luciferase reporter gene assay and chromatin immunoprecipitation (ChIP) assay. The anti-tumor effects and the potential toxic side effects of CPP alone or in combination with TRAIL were also evaluated in vivo. -

XIAP's Profile in Human Cancer
biomolecules Review XIAP’s Profile in Human Cancer Huailu Tu and Max Costa * Department of Environmental Medicine, Grossman School of Medicine, New York University, New York, NY 10010, USA; [email protected] * Correspondence: [email protected] Received: 16 September 2020; Accepted: 25 October 2020; Published: 29 October 2020 Abstract: XIAP, the X-linked inhibitor of apoptosis protein, regulates cell death signaling pathways through binding and inhibiting caspases. Mounting experimental research associated with XIAP has shown it to be a master regulator of cell death not only in apoptosis, but also in autophagy and necroptosis. As a vital decider on cell survival, XIAP is involved in the regulation of cancer initiation, promotion and progression. XIAP up-regulation occurs in many human diseases, resulting in a series of undesired effects such as raising the cellular tolerance to genetic lesions, inflammation and cytotoxicity. Hence, anti-tumor drugs targeting XIAP have become an important focus for cancer therapy research. RNA–XIAP interaction is a focus, which has enriched the general profile of XIAP regulation in human cancer. In this review, the basic functions of XIAP, its regulatory role in cancer, anti-XIAP drugs and recent findings about RNA–XIAP interactions are discussed. Keywords: XIAP; apoptosis; cancer; therapeutics; non-coding RNA 1. Introduction X-linked inhibitor of apoptosis protein (XIAP), also known as inhibitor of apoptosis protein 3 (IAP3), baculoviral IAP repeat-containing protein 4 (BIRC4), and human IAPs like protein (hILP), belongs to IAP family which was discovered in insect baculovirus [1]. Eight different IAPs have been isolated from human tissues: NAIP (BIRC1), BIRC2 (cIAP1), BIRC3 (cIAP2), XIAP (BIRC4), BIRC5 (survivin), BIRC6 (apollon), BIRC7 (livin) and BIRC8 [2]. -

Expression of the Tumor Necrosis Factor Receptor-Associated Factors
Expression of the Tumor Necrosis Factor Receptor- Associated Factors (TRAFs) 1 and 2 is a Characteristic Feature of Hodgkin and Reed-Sternberg Cells Keith F. Izban, M.D., Melek Ergin, M.D, Robert L. Martinez, B.A., HT(ASCP), Serhan Alkan, M.D. Department of Pathology, Loyola University Medical Center, Maywood, Illinois the HD cell lines. Although KMH2 showed weak Tumor necrosis factor receptor–associated factors expression, the remaining HD cell lines also lacked (TRAFs) are a recently established group of proteins TRAF5 protein. These data demonstrate that consti- involved in the intracellular signal transduction of tutive expression of TRAF1 and TRAF2 is a charac- several members of the tumor necrosis factor recep- teristic feature of HRS cells from both patient and tor (TNFR) superfamily. Recently, specific members cell line specimens. Furthermore, with the excep- of the TRAF family have been implicated in promot- tion of TRAF1 expression, HRS cells from the three ing cell survival as well as activation of the tran- HD cell lines showed similar TRAF protein expres- scription factor NF- B. We investigated the consti- sion patterns. Overall, these findings demonstrate tutive expression of TRAF1 and TRAF2 in Hodgkin the expression of several TRAF proteins in HD. Sig- and Reed–Sternberg (HRS) cells from archived nificantly, the altered regulation of selective TRAF paraffin-embedded tissues obtained from 21 pa- proteins may reflect HRS cell response to stimula- tients diagnosed with classical Hodgkin’s disease tion from the microenvironment and potentially (HD). In a selective portion of cases, examination of contribute both to apoptosis resistance and cell HRS cells for Epstein-Barr virus (EBV)–encoded maintenance of HRS cells. -
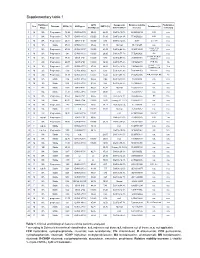
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

Beyond Tumor Necrosis Factor Receptor: TRADD Signaling in Toll-Like Receptors
Beyond tumor necrosis factor receptor: TRADD signaling in toll-like receptors Nien-Jung Chen*†‡, Iok In Christine Chio*‡, Wen-Jye Lin*, Gordon Duncan*, Hien Chau*§, David Katz*¶, Huey-Lan Huang*ʈ, Kelly A. Pike*,**, Zhenyue Hao*, Yu-Wen Su*, Kazuo Yamamoto*, Rene´ e F. de Pooter††, Juan Carlos Zu´ n˜ iga-Pflu¨ cker††, Andrew Wakeham*, Wen-Chen Yeh*, and Tak W. Mak*‡‡ *The Campbell Family Institute for Breast Cancer Research, Ontario Cancer Institute, University Health Network and Department of Medical Biophysics, University of Toronto, Toronto, ON, Canada M5G 2C1; †Institute of Microbiology and Immunology, School of Life Science, National Yang-Ming University, Taipei, Taiwan 112, Republic of China; and ††Department of Immunology, University of Toronto, Sunnybrook and Women’s College Health Sciences Centre, 2075 Bayview Avenue, Toronto, ON, Canada M4N 3M5 Contributed by Tak W. Mak, July 9, 2008 (sent for review June 24, 2008) Tumor necrosis factor receptor 1-associated death domain protein tory responses in vivo, but also that this molecule is involved in (TRADD) is the core adaptor recruited to TNF receptor 1 (TNFR1) germinal center (GC) formation, T cell costimulation, and TLR upon TNF␣ stimulation. In cells from TRADD-deficient mice, TNF␣- signaling. Our TRADD knockout mice represent a very useful mediated apoptosis and TNF␣-stimulated NF-B, JNK, and ERK tool for extending the ever-increasing list of TRADD functions activation are defective. TRADD is also important for germinal in vitro and in vivo. center formation, DR3-mediated costimulation of T cells, and TNF␣- mediated inflammatory responses in vivo. TRADD deficiency does Results not enhance IFN␥-induced signaling. -

Nuclear TRAF3 Is a Negative Regulator of CREB in B Cells
Nuclear TRAF3 is a negative regulator of CREB in B cells Nurbek Mambetsarieva,b,c,1, Wai W. Linb,1, Laura L. Stunza,d, Brett M. Hansona, Joanne M. Hildebranda,2, and Gail A. Bishopa,b,d,e,f,3 aDepartment of Microbiology, University of Iowa, Iowa City, IA 52242; bImmunology Graduate Program, University of Iowa, Iowa City, IA 52242; cMedical Scientist Training Program, University of Iowa, Iowa City, IA 52242; dHolden Comprehensive Cancer Center, University of Iowa, Iowa City, IA 52242; eInternal Medicine, University of Iowa, Iowa City, IA 52242; and fDepartment of Veterans Affairs Medical Center, Research (151), Iowa City, IA 52246 Edited by Louis M. Staudt, National Cancer Institute, NIH, Bethesda, MD, and approved December 14, 2015 (received for review July 23, 2015) The adaptor protein TNF receptor-associated factor 3 (TRAF3) regu- deficient T cells and macrophages lack the enhanced survival phe- lates signaling through B-lymphocyte receptors, including CD40, notype, although they display constitutive NF-κB2 activation (12, BAFF receptor, and Toll-like receptors, and also plays a critical role 13). TRAF3 degradation is neither necessary nor sufficient for B-cell inhibiting B-cell homoeostatic survival. Consistent withthesefindings, NF-κB2 activation (14). These findings indicate that TRAF3 regu- loss-of-function human TRAF3 mutations are common in B-cell cancers, lates additional important prosurvival pathways in B cells. particularly multiple myeloma and B-cell lymphoma. B cells of B-cell– Nuclear localization of TRAF3 has been reported in several specific TRAF3−/− mice (B-Traf3−/−) display remarkably enhanced sur- nonhematopoietic cell types (15, 16), but the function of TRAF3 vival compared with littermate control (WT) B cells. -

Essential Role of Survivin, an Inhibitor of Apoptosis Protein, in T Cell
Essential Role of Survivin, an Inhibitor of Apoptosis Protein, in T Cell Development, Maturation, and Homeostasis Zheng Xing,1 Edward M. Conway,2 Chulho Kang,1 and Astar Winoto1 1Department of Molecular and Cell Biology, Division of Immunology and Cancer Research Laboratory, University of California at Berkeley, Berkeley, CA 94720 2Center for Transgene Technology and Gene Therapy, Flanders Interuniversity Institute for Biotechnology, University of Leuven, B-3000 Leuven, Belgium Abstract Survivin is an inhibitor of apoptosis protein that also functions during mitosis. It is expressed in all common tumors and tissues with proliferating cells, including thymus. To examine its role in apoptosis and proliferation, we generated two T cell–specific survivin-deficient mouse lines with deletion occurring at different developmental stages. Analysis of early deleting survivin mice showed arrest at the pre–T cell receptor proliferating checkpoint. Loss of survivin at a later stage resulted in normal thymic development, but peripheral T cells were immature and significantly reduced in number. In contrast to in vitro studies, loss of survivin does not lead to increased apoptosis. However, newborn thymocyte homeostatic and mitogen-induced proliferation of survivin-deficient T cells were greatly impaired. These data suggest that survivin is not essential for T cell apoptosis but is crucial for T cell maturation and proliferation, and survivin-mediated homeostatic expansion is an important physiological process of T cell development. Key words: proliferation • apoptosis • T cell development • survivin • IAP Introduction The thymus is the major organ of T lymphocyte maturation CD4 CD8 or CD4 CD8 (single positive [SP]) cells. and differentiation. During development, T cells have to These mature cells then migrate to the peripheral immune confront sequentially fateful decisions: the pre-TCR organs where they carry out their major function in defend- checkpoint, TCR chain rearrangements, positive selection, ing the body against foreign invasion. -

Survivin-3B Potentiates Immune Escape in Cancer but Also Inhibits the Toxicity of Cancer Chemotherapy
Published OnlineFirst July 15, 2013; DOI: 10.1158/0008-5472.CAN-13-0036 Cancer Molecular and Cellular Pathobiology Research Survivin-3B Potentiates Immune Escape in Cancer but Also Inhibits the Toxicity of Cancer Chemotherapy Fred erique Vegran 1,5, Romain Mary1, Anne Gibeaud1,Celine Mirjolet2, Bertrand Collin4,6, Alexandra Oudot4, Celine Charon-Barra3, Laurent Arnould3, Sarab Lizard-Nacol1, and Romain Boidot1 Abstract Dysregulation in patterns of alternative RNA splicing in cancer cells is emerging as a significant factor in cancer pathophysiology. In this study, we investigated the little known alternative splice isoform survivin-3B (S-3B) that is overexpressed in a tumor-specific manner. Ectopic overexpression of S-3B drove tumorigenesis by facilitating immune escape in a manner associated with resistance to immune cell toxicity. This resistance was mediated by interaction of S-3B with procaspase-8, inhibiting death-inducing signaling complex formation in response to Fas/ Fas ligand interaction. We found that S-3B overexpression also mediated resistance to cancer chemotherapy, in this case through interactions with procaspase-6. S-3B binding to procaspase-6 inhibited its activation despite mitochondrial depolarization and caspase-3 activation. When combined with chemotherapy, S-3B targeting in vivo elicited a nearly eradication of tumors. Mechanistic investigations identified a previously unrecognized 7-amino acid region as responsible for the procancerous properties of survivin proteins. Taken together, our results defined S-3B as an important functional actor in tumor formation and treatment resistance. Cancer Res; 73(17); 1–11. Ó2013 AACR. Introduction that can induce the expression of five different transcripts with D Alternative splicing is an important mechanism for the different functions: survivin, survivin- Ex3, survivin-2B (5), a generation of the variety of proteins indispensable for cell survivin-3B (S-3B; ref. -

Synthetic Bichalcone TSWU-BR23 Induces Apoptosis of Human Colon Cancer HT-29 Cells by P53-Mediated Mitochondrial Oligomerization
ANTICANCER RESEARCH 35: 5407-5416 (2015) Synthetic Bichalcone TSWU-BR23 Induces Apoptosis of Human Colon Cancer HT-29 Cells by p53-Mediated Mitochondrial Oligomerization of BAX/BAK and Lipid Raft Localization of CD95/FADD MENG-LIANG LIN1, SHIH-SHUN CHEN2 and TIAN-SHUNG WU3 1Department of Medical Laboratory Science and Biotechnology, China Medical University, Taichung, Taiwan, R.O.C.; 2Department of Medical Laboratory Science and Biotechnology, Central Taiwan University of Science and Technology, Taichung, Taiwan, R.O.C.; 3Department of Chemistry, National Cheng Kung University, Tainan, Taiwan, R.O.C. Abstract. A synthetic bichalcone analog, (E)-1-(3-((4-(4- Apoptosis is a key regulatory mechanism of tissue and acetylphenyl)piperazin-1-yl)methyl)-4-hydroxy-5- cellular homeostasis that can be induced via activation of methoxyphenyl)-3-(pyridin-3-yl)prop-2-en-1-one (TSWU- either the extrinsic or the intrinsic signaling pathway (1). The BR23), has been shown to induce apoptosis in human colon extrinsic pathway is triggered by binding cluster of cancer HT-29 cells involving the induction of CD95 and FAS- differentiation 95 (CD95) ligand to CD95 receptor on the associated protein death domain (FADD), but its precise target cells, which in turn leads to recruitment of cytosolic mechanism of action has not been fully elucidated. Using adaptor FAS-associated death domain-containing protein cell-surface biotinylation and sucrose density-gradient-based (FADD) and pro-caspase-8. Upon co-localization with membrane flotation techniques, we showed that the disruption FADD, pro-caspase-8 undergoes auto-proteolytic cleavage, of TSWU-BR23-induced lipid raft localization of CD95/FADD releasing activated caspase-8 into the cytosol, where it causes by cholesterol-depleting agent (methyl-β-cyclodextrin) was cleavage of BH3-interacting domain death agonist (BID) into reversed by cholesterol replenishment.