KULAGE-DISSERTATION-2013.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Concurrent Reduction and Distillation; an Improved Technique for the Recovery and Chemical Refinement of the Isotopes of Cadmium and Zinc*
If I CONCURRENT REDUCTION AND DISTILLATION; AN IMPROVED TECHNIQUE FOR THE RECOVERY AND CHEMICAL REFINEMENT OF THE ISOTOPES OF CADMIUM AND ZINC* H. H. Cardill L. E. McBride LOHF-821046—2 E. W. McDaniel DE83 001931 Operations Division Oak Ridge National Laboratory Oak Ridge, Tennessee 3 7830 .DISCLAIMER • s prepared es an accouni oi WOTk sconsQ'ed by an agency of itie United Slates Cover r "iiit?3 Stilt 65 Gov(?fnft»gn ^ ri(jt ^nv aQcncy Tllf GO* ^or rlny O^ tt^^ir ^rnployut^. TigV MASTER lostifl. ion, apod'aiuj pioiJut;. <y vned nghtv Reference hcein xc any v>t^-'T*t. ^ Dv ^.r^de narno, tf^Oernafk^ mir'ij'iJClureT. or otrierw «d <Jo*^ •nenaalion. or liivONng bv 'Vhe United _^_ . vs and opinions of authors enp'«i«] herein do noi IOM t' irip UnitK* Stales Govefnrneni c a^v agency For presentation at the 11th World Conference of the International Nuclear Target Development Society, October 6-8, 1982, Seattle, Washington. By acceptance of this article, the publisher or recipient acknowledge* the U.S. Govarnment's right to retain a nonaxclusiva, royalty-frtta license in and to any copyright covering tha article. NOTICE P0T09_0£-Jfns,J^SPQRT_JfgE_Il,LESIBLE. I has been reproauce.1 from the best available aopy to pensit the broadest possible avail- ability* *Research sponsored by the Office of Basic Energy Science, U.S. Department of Energy under contract W-7405-eng-26 with the Union Carbide Co rpcration,, DISTRIBUTION OF THIS DOCUMEHT 18 UNLIMITED ' CONCURRENT REDUCTION AND DISTILLATION - AY. IMPROVED TECHNIQUE FOR THE RECOVERY AND CHEMICAL REFINEMENT OF THE ISOTOPES OF CADMIUM AND ZINC H. -

Neutron Capture Cross Sections of Cadmium Isotopes
Neutron Capture Cross Sections of Cadmium Isotopes By Allison Gicking A thesis submitted to Oregon State University In partial fulfillment of the requirements for the degree of Bachelor of Science Presented June 8, 2011 Commencement June 17, 2012 Abstract The neutron capture cross sections of 106Cd, 108Cd, 110Cd, 112Cd, 114Cd and 116Cd were determined in the present project. Four different OSU TRIGA reactor facilities were used to produce redundancy in the results and to measure the thermal cross section and resonance integral separately. When the present values were compared with previously measured values, the differences were mostly due to the kind of detector used or whether or not the samples were natural cadmium. Some of the isotopes did not have any previously measured values, and in that case, new information about the cross sections of those cadmium isotopes has been provided. Table of Contents I. Introduction………………………………………………………………….…….…1 II. Theory………………………………………………………………………...…...…3 1. Neutron Capture…………………………………………………….….……3 2. Resonance Integral vs. Effective Thermal Cross Section…………...………5 3. Derivation of the Activity Equations…………………………………....…..8 III. Methods………………………………………………………….................…...…...12 1. Irradiation of the Samples………………………………………….….....…12 2. Sample Preparation and Parameters………………..………...………..……16 3. Efficiency Calibration of Detectors…………………………..………....…..18 4. Data Analysis…………………………………...…….………………...…..19 5. Absorption by 113Cd……………………………………...……...….………20 IV. Results………………………………………………….……………..……….…….22 -
![Arxiv:1305.1738V1 [Nucl-Ex] 8 May 2013 Ae Pcrsoystpa SLECR.High-Energy ISOLDE-CERN](https://docslib.b-cdn.net/cover/6428/arxiv-1305-1738v1-nucl-ex-8-may-2013-ae-pcrsoystpa-slecr-high-energy-isolde-cern-1276428.webp)
Arxiv:1305.1738V1 [Nucl-Ex] 8 May 2013 Ae Pcrsoystpa SLECR.High-Energy ISOLDE-CERN
Spins, Electromagnetic Moments, and Isomers of 107-129Cd D. T. Yordanov,1,2, ∗ D. L. Balabanski,3 J. Biero´n,4 M. L. Bissell,5 K. Blaum,1 I. Budinˇcevi´c,5 S. Fritzsche,6 N. Fr¨ommgen,7 G. Georgiev,8 Ch. Geppert,6, 7 M. Hammen,7 M. Kowalska,2 K. Kreim,1 A. Krieger,7 R. Neugart,7 W. N¨ortersh¨auser,6, 7 J. Papuga,5 and S. Schmidt6 1Max-Planck-Institut f¨ur Kernphysik, Saupfercheckweg 1, D-69117 Heidelberg, Germany 2CERN European Organization for Nuclear Research, Physics Department, CH-1211 Geneva 23, Switzerland 3INRNE, Bulgarian Academy of Science, BG-1784 Sofia, Bulgaria 4Instytut Fizyki imienia Mariana Smoluchowskiego, Uniwersytet Jagiello´nski, Reymonta 4, 30-059 Krak´ow, Poland 5Instituut voor Kern- en Stralingsfysica, KU Leuven, Celestijnenlaan 200D, B-3001 Leuven, Belgium 6GSI Helmholtzzentrum f¨ur Schwerionenforschung GmbH, D-64291 Darmstadt, Germany 7Institut f¨ur Kernchemie, Johannes Gutenberg-Universit¨at Mainz, D-55128 Mainz, Germany 8CSNSM-IN2P3-CNRS, Universit´ede Paris Sud, F-91405 Orsay, France (Dated: October 7, 2018) The neutron-rich isotopes of cadmium up to the N = 82 shell closure have been investigated by high-resolution laser spectroscopy. Deep-UV excitation at 214.5 nm and radioactive-beam bunching provided the required experimental sensitivity. Long-lived isomers are observed in 127Cd and 129Cd for the first time. One essential feature of the spherical shell model is unambiguously confirmed by − a linear increase of the 11/2 quadrupole moments. Remarkably, this mechanism is found to act well beyond the h11/2 shell. PACS numbers: 21.10.Ky, 21.60.Cs, 32.10.Fn, 31.15.aj When first proposed the nuclear shell model was protons impinging on a tungsten rod produced low- to largely justified on the basis of magnetic-dipole proper- medium-energy neutrons inducing fission in a uranium ties of nuclei [1]. -

Atomic Weights of the Elements 2013 (IUPAC Technical Report)
Pure Appl. Chem. 2016; 88(3): 265–291 IUPAC Technical Report Juris Meija*, Tyler B. Coplen, Michael Berglund, Willi A. Brand, Paul De Bièvre, Manfred Gröning, Norman E. Holden, Johanna Irrgeher, Robert D. Loss, Thomas Walczyk and Thomas Prohaska Atomic weights of the elements 2013 (IUPAC Technical Report) DOI 10.1515/pac-2015-0305 Received March 26, 2015; accepted December 8, 2015 Abstract: The biennial review of atomic-weight determinations and other cognate data has resulted in changes for the standard atomic weights of 19 elements. The standard atomic weights of four elements have been revised based on recent determinations of isotopic abundances in natural terrestrial materials: cadmium to 112.414(4) from 112.411(8), molybdenum to 95.95(1) from 95.96(2), selenium to 78.971(8) from 78.96(3), and thorium to 232.0377(4) from 232.038 06(2). The Commission on Isotopic Abundances and Atomic Weights (ciaaw.org) also revised the standard atomic weights of fifteen elements based on the 2012 Atomic Mass Evaluation: aluminium (aluminum) to 26.981 5385(7) from 26.981 5386(8), arsenic to 74.921 595(6) from 74.921 60(2), beryllium to 9.012 1831(5) from 9.012 182(3), caesium (cesium) to 132.905 451 96(6) from 132.905 4519(2), cobalt to 58.933 194(4) from 58.933 195(5), fluorine to 18.998 403 163(6) from 18.998 4032(5), gold to 196.966 569(5) from 196.966 569(4), holmium to 164.930 33(2) from 164.930 32(2), manganese to 54.938 044(3) from 54.938 045(5), niobium to 92.906 37(2) from 92.906 38(2), phosphorus to 30.973 761 998(5) from 30.973 762(2), praseodymium to 140.907 66(2) from 140.907 65(2), Article note: Sponsoring body: IUPAC Inorganic Chemistry Division Committee: see more details on page 289. -

Precision Mass Measurements for Studies of Nucleosynthesis Via the Rapid Neutron-Capture Process
Dissertation submitted to the Combined Faculties for the Natural Sciences and for Mathematics of the Ruperto-Carola University of Heidelberg, Germany for the degree of Doctor of Natural Sciences Put forward by Diplomant: Dinko Atanasov Born in: Yambol, Bulgaria Oral examination: 06. July 2016 Precision mass measurements for studies of nucleosynthesis via the rapid neutron-capture process Penning-trap mass measurements of neutron-rich cadmium and caesium isotopes Prof. Dr. Klaus Blaum Referees: PD Dr. Adriana P´alffy Zusammenfasung Obwohl die Theorie ¨uber die schnelle Neutronenanlagerung (r-Prozess) schon vor mehr als 55 Jahren entwickelt wurde, wird ¨uber den exakten Ort im Universum an dem dieser Prozess stattfindet noch rege debattiert. Theoretische Studien sagen voraus, dass der Verlauf des r-Prozesses gepr¨agtwird durch ¨außerstneutronenreiche Materie mit sehr asymmetrischen Proton-zu-Neutron Verh¨altnissen.Die aktuellen Kenntnisse ¨uber Eigenschaften dieser neutro- nenreichen Isotope, die dazu geeignet sind als Eingangsdaten f¨urStudien ¨uber den r-Prozess herangezogen zu werden, sind nur unzureichend oder gar nicht vorhanden. Die grundlegen- den Eigenschaften der Kerne, wie Bindungsenergie, Halbwertszeiten und Reaktionsquerschnitt spielen eine wichtige Rolle in den theoretischen Simulationen und k¨onnendiese beeinflussen oder sogar zu drastisch alternativen Ergebnissen f¨uhren. Um diese Theorien mit gemesse- nen Daten zu untermauern und die Produktion neutronenreicher Isotope zu verbessern, wurde an Forschungseinrichtungen wie ISOLDE am CERN, ein g¨anzlich bemerkenswerter Aufwand betrieben. Das Ziel dieser Dissertation ist es die experimentelle Arbeit zu beschreiben, welche n¨otigwar, um die Pr¨azisionsmassenmessungender neutronenreichen Isotope Cadmium (129−131Cd) und C¨asium(132;146−148Cs) zu erm¨oglichen. Die Messungen wurden an der Iso- topenfabrik ISOLDE am CERN mithilfe des aus vier Ionenfallen bestehenden Massenspek- trometers ISOLTRAP durchgef¨uhrt. -

Stable Isotope Separation in Calutrons
Printed ill tho llnitzd Statss ot 4mcm 4vailsble frurii Nations' Technical Information Service U S :3ep?rtmelli uf Commerce 5285 Port 4oyal Cosd Spririizflald Viryinis 22161 NTlS ptwiodes P~III~~Copy ,498, Microfiche P,C1 I his rcpoti \*!is prspxed as an account of >,dSi-k sponsored by a3 agsncy of the United Sraies Governr;,ent. Neither thc !J nited Skiics i<n.~cti $IK~: nor any agency th2reof. no? any of thzir employoes, (ciakc?.Aijy ivarranty, express or irripiled, or assunias any legal liability or responsibility for ihe accuracy. compleienass, or usefulness of any inforriraiton. ;p ius, product, or process disclosed, or represents that its usewould not infr privately o-v?d ilghts Rcferencc herein to ai-y specific commercial product, process, or service by trade nziile, Zi'adei*lark, manufacturer, or otherwise, does not necsssarily constitute or imply its enduisemcat, recorn,n faVOiii-,g by ille United States(;ovc;riiiie!ii 01- any Zgency thereof Th opinions of auiihols cxp:essed here:!: d~ ~iui neccssarily statc or refisc?!host of the United Sta?ezGovernmer:?or any ngcncy therccf. ____ ___________... ..... .____ ORNL/TM-10356 OPERATIONS DIVISION STABLE ISOTOPE SEPARATION IN CALUTRONS: FORTY YEARS OF PRODUCTION AND DISTRIBUTION W. A. Bell J. G. Tracy Date Published: November 1987 Prepared by the OAK RIDGE NATIONAL LABORATORY Oak Ridge, Tennessee 37831 operated by MARTIN MARIETTA ENERGY SYSTEMS, INC. for the U.S. DEPARTMENT OF ENERGY under Contract No. DE-AC05-840R21400 iti CONTENTS Page ACKNOWLEDGMENTS ................................................. iv I. INTRODUCTION .............................................. 1 11. PROGRAM EVOLUTION ......................................... 1 111. RESEARCH AND DEVELOPMENT .................................. 5 IV. CHEMISTRY ................................................. 8 V. -

4.48 Cadmium
IUPAC 4.48 cadmium Stable Relative Mole isotope atomic mass fraction 106 Cd 105.906 460 0.012 45 108 Cd 107.904 183 0.008 88 110 Cd 109.903 007 0.124 70 111 Cd 110.904 183 0.127 95 112 Cd 111.902 763 0.241 09 113 † Cd 112.904 408 0.122 27 114 Cd 113.903 365 0.287 54 116 † Cd 115.904 763 0.075 12 † Radioactive isotope having a relatively long half-life and a characteristic terrestrial isotopic composition that contributes significantly and reproducibly to the determination of the standard atomic weight of the element in normal materials . Half-lives of 113 Cd and 116 Cd are 8.04 × 10 15 years and 3 × 10 19 years, respectively. P.O. 13757, Research Triangle Park, NC (919) 485-8700 IUPAC 4.48.1 Cadmium isotopes in biology Metal accumulation is a threat to our world’s water systems and wildlife. As a way to measure the influence of heavy metals on wildlife with mass spectrometric techniques, some researchers use animal food enriched in specific cadmium isotopes . These experiments work by exposing the animals to a diet enriched in 106 Cd and/or other stable isotopes of metals (for example, 65 Cu and/or 62 Ni) for a period of time. Depending on the purpose of the experiment, the residence time of the food in the gut is determined and isotopic compositions of the gut and/or feces are measured via inductively coupled plasma mass spectrometry (ICP-MS). This information is used to measure bio-uptake (absorption and incorporation of a substance by living tissue) and accumulation rates of metals in an exposed animal [352, 353]. -
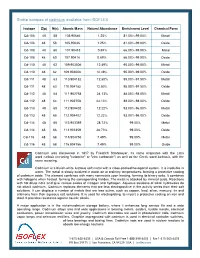
Stable Isotopes of Cadmium Available from ISOFLEX
Stable isotopes of cadmium available from ISOFLEX Isotope Z(p) N(n) Atomic Mass Natural Abundance Enrichment Level Chemical Form Cd-106 48 58 105.90646 1.25% 81.00%-99.00% Metal Cd-106 48 58 105.90646 1.25% 81.00%-99.00% Oxide Cd-108 48 60 107.90418 0.89% 66.00%-99.00% Metal Cd-108 48 60 107.90418 0.89% 66.00%-99.00% Oxide Cd-110 48 62 109.903006 12.49% 95.00%-99.00% Metal Cd-110 48 62 109.903006 12.49% 95.00%-99.00% Oxide Cd-111 48 63 110.904182 12.80% 95.00%-97.50% Metal Cd-111 48 63 110.904182 12.80% 95.00%-97.50% Oxide Cd-112 48 64 111.902758 24.13% 88.00%-98.00% Metal Cd-112 48 64 111.902758 24.13% 88.00%-98.00% Oxide Cd-113 48 65 112.904402 12.22% 93.00%-96.00% Metal Cd-113 48 65 112.904402 12.22% 93.00%-96.00% Oxide Cd-114 48 66 113.903359 28.73% 99.00% Metal Cd-114 48 66 113.903359 28.73% 99.00% Oxide Cd-116 48 68 115.904756 7.49% 99.00% Metal Cd-116 48 68 115.904756 7.49% 99.00% Oxide Cadmium was discovered in 1817 by Friedrich Strohmeyer. Its name originates with the Latin word cadmia (meaning "calamine" or "zinc carbonate") as well as the Greek word kadmeia, with the same meaning. Cadmium is a bluish-white lustrous soft metal with a close-packed hexagonal system. -

Atomic Weights of the Elements 2013 (IUPAC Technical Report)
NRC Publications Archive Archives des publications du CNRC Atomic weights of the elements 2013 (IUPAC Technical Report) Meija, Juris; Coplen, Tyler B.; Berglund, Michael; Brand, Willi A.; De Bièvre, Paul; Gröning, Manfred; Holden, Norman E.; Irrgeher, Johanna; Loss, Robert D.; Walczyk, Thomas; Prohaska, Thomas This publication could be one of several versions: author’s original, accepted manuscript or the publisher’s version. / La version de cette publication peut être l’une des suivantes : la version prépublication de l’auteur, la version acceptée du manuscrit ou la version de l’éditeur. For the publisher’s version, please access the DOI link below./ Pour consulter la version de l’éditeur, utilisez le lien DOI ci-dessous. Publisher’s version / Version de l'éditeur: https://doi.org/10.1515/pac-2015-0305 Pure and Applied Chemistry, 88, 3, pp. 265-291, 2016-02-24 NRC Publications Record / Notice d'Archives des publications de CNRC: https://nrc-publications.canada.ca/eng/view/object/?id=23a71e6c-605b-458b-b56e-5efb0a347895 https://publications-cnrc.canada.ca/fra/voir/objet/?id=23a71e6c-605b-458b-b56e-5efb0a347895 Access and use of this website and the material on it are subject to the Terms and Conditions set forth at https://nrc-publications.canada.ca/eng/copyright READ THESE TERMS AND CONDITIONS CAREFULLY BEFORE USING THIS WEBSITE. L’accès à ce site Web et l’utilisation de son contenu sont assujettis aux conditions présentées dans le site https://publications-cnrc.canada.ca/fra/droits LISEZ CES CONDITIONS ATTENTIVEMENT AVANT D’UTILISER CE SITE WEB. Questions? Contact the NRC Publications Archive team at [email protected]. -
Cyclotron Produced Radionuclides: Operation and Maintenance of Gas and Liquid Targets
IAEA RADIOISOTOPES AND RADIOPHARMACEUTICALS SERIES No. 4 This publication, which draws on the results of an IAEA coordinated MAINTENANCEAND GASOF TARGETS LIQUID AND OPERATION RADIONUCLIDES: PRODUCED CYCLOTRON research project and on the input from dedicated experts in the fi eld, provides a comprehensive overview of the technologies involved in the manufacturing and operation of liquid and gas targets for cyclotron based production of radioisotopes. It covers the technology behind targetry, techniques for preparation of targets, irradiation of targets under high beam currents, target processing, target recovery, etc. The publication will be useful to scientists and technologists interested in translating cyclotron based radioisotope production into practice, as well as to postgraduate students in the fi eld. Cyclotron Produced Radionuclides: Operation and Maintenance of Gas and Liquid Targets INTERNATIONAL ATOMIC ENERGY AGENCY VIENNA ISBN 978–92–0–130710–1 ISSN 2077–6462 RELATED PUBLICATIONS IAEA RADIOISOTOPES AND RADIOPHARMACEUTICALS SERIES PUBLICATIONS TECHNETIUM-99m RADIOPHARMACEUTICALS: STATUS AND TRENDS IAEA Radioisotopes and Radiopharmaceuticals Series No. 1 STI/PUB/1405 (360 pp.; 2009) One of the main objectives of the IAEA Radioisotope Production and Radiation Technology programme is to enhance the expertise and capability of IAEA Member States in deploying ISBN 978–92–0–103509–7 Price: €52.00 emerging radioisotope products and generators for medical and industrial applications in order to meet national needs as well as to assimilate new developments in radiopharmaceuticals for PRODUCTION OF LONG LIVED PARENT RADIONUCLIDES 68 82 90 188 diagnostic and therapeutic applications. This will ensure local availability of these applications FOR GENERATORS: Ge, Sr, Sr AND W within a framework of quality assurance. -
Radiological and Chemical Fact Sheets to Support Health Risk Analyses for Contaminated Areas
Radiological and Chemical Fact Sheets to Support Health Risk Analyses for Contaminated Areas Prepared by Argonne National Laboratory Environmental Science Division John Peterson, Margaret MacDonell, Lynne Haroun, and Fred Monette In collaboration with U.S. Department of Energy Richland Operations Office R. Douglas Hildebrand and Chicago Operations Office Anibal Taboas March 2007 Radiological and Chemical Fact Sheets to Support Health Risk Analyses for Contaminated Areas These fact sheets summarize health-related information for contaminants present in the environment as a result of past industrial activities and other releases. The objective is to provide scientific context for risk analyses to guide health protection measures. Geared toward an audience familiar with basic risk concepts, they were originally developed for the U.S. Department of Energy (DOE) Richland and Chicago Operations Offices to serve as an information resource for people involved in environmental programs. The initial set was expanded to address evolving homeland security concerns, and these 51 radiological and chemical fact sheets also serve as a scientific information resource for the public. Twenty-nine radionuclide-specific fact sheets have been prepared: ¾ Americium ¾ Iridium ¾ Selenium ¾ Cadmium ¾ Krypton ¾ Strontium ¾ Californium ¾ Neptunium ¾ Technetium ¾ Carbon-14 ¾ Nickel ¾ Thorium ¾ Cesium ¾ Plutonium ¾ Tin ¾ Chlorine ¾ Polonium ¾ Tritium ¾ Cobalt ¾ Potassium-40 ¾ Uranium ¾ Curium ¾ Protactinium ¾ Depleted uranium (DU) ¾ Europium ¾ Radium complements uranium -

Decay Spectroscopy of Neutron-Rich Cadmium Around the N = 82 Shell Closure
Decay Spectroscopy of Neutron-Rich Cadmium Around the N = 82 Shell Closure by Nikita Bernier B.Sc., Universit´eLaval, 2011 M.Sc., Universit´eLaval, 2013 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate and Postdoctoral Studies (Physics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) December 2018 c Nikita Bernier 2018 The following individuals certify that they have read, and recommend to the Faculty of Graduate and Postdoctoral Studies for acceptance, the dissertation entitled: Decay Spectroscopy of Neutron-Rich Cadmium Around the N = 82 Shell Closure submitted by Nikita Bernier in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Physics. Examining Committee: Dr Reiner Kr¨ucken, Physics Supervisor Dr Colin Gay, Physics Supervisory Committee Member Dr Janis McKenna, Physics University Examiner Dr Chris Orvig, Chemistry University Examiner Additional Supervisory Committee Members: Dr Sonia Bacca, Physics Supervisory Committee Member Dr Robert Kiefl, Physics Supervisory Committee Member ii Abstract The neutron-rich cadmium isotopes (Z = 49) near the well-known magic numbers at Z = 50 and N = 82 are prime candidates to study the evolving shell structure observed in exotic nuclei. Additionally, nuclei around the doubly-magic 132Sn have been demonstrated to have direct implications for astrophysical models, leading to the r-process abundance peak at A ≈ 130 and the corresponding waiting-point nuclei around N = 82. The β-decay of the N = 82 isotope 130Cd into 130In was investigated in 2002 [1], but the information for states of the lighter indium isotope 128In is still limited.