Machine Learning with Digital Signal Processing for Ultrafast, Accurate, and Scalable Genome Classification at All Taxonomic Levels
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ML-DSP: Machine Learning with Digital Signal Processing for Ultrafast, Accurate, and Scalable Genome Classification at All Taxonomic Levels Gurjit S
Randhawa et al. BMC Genomics (2019) 20:267 https://doi.org/10.1186/s12864-019-5571-y SOFTWARE ARTICLE Open Access ML-DSP: Machine Learning with Digital Signal Processing for ultrafast, accurate, and scalable genome classification at all taxonomic levels Gurjit S. Randhawa1* , Kathleen A. Hill2 and Lila Kari3 Abstract Background: Although software tools abound for the comparison, analysis, identification, and classification of genomic sequences, taxonomic classification remains challenging due to the magnitude of the datasets and the intrinsic problems associated with classification. The need exists for an approach and software tool that addresses the limitations of existing alignment-based methods, as well as the challenges of recently proposed alignment-free methods. Results: We propose a novel combination of supervised Machine Learning with Digital Signal Processing, resulting in ML-DSP: an alignment-free software tool for ultrafast, accurate, and scalable genome classification at all taxonomic levels. We test ML-DSP by classifying 7396 full mitochondrial genomes at various taxonomic levels, from kingdom to genus, with an average classification accuracy of > 97%. A quantitative comparison with state-of-the-art classification software tools is performed, on two small benchmark datasets and one large 4322 vertebrate mtDNA genomes dataset. Our results show that ML-DSP overwhelmingly outperforms the alignment-based software MEGA7 (alignment with MUSCLE or CLUSTALW) in terms of processing time, while having comparable classification accuracies for small datasets and superior accuracies for the large dataset. Compared with the alignment-free software FFP (Feature Frequency Profile), ML-DSP has significantly better classification accuracy, and is overall faster. We also provide preliminary experiments indicating the potential of ML-DSP to be used for other datasets, by classifying 4271 complete dengue virus genomes into subtypes with 100% accuracy, and 4,710 bacterial genomes into phyla with 95.5% accuracy. -

Employing Geographical Information Systems in Fisheries Management in the Mekong River: a Case Study of Lao PDR
Employing Geographical Information Systems in Fisheries Management in the Mekong River: a case study of Lao PDR Kaviphone Phouthavongs A thesis submitted in partial fulfilment of the requirement for the Degree of Master of Science School of Geosciences University of Sydney June 2006 ABSTRACT The objective of this research is to employ Geographical Information Systems to fisheries management in the Mekong River Basin. The study uses artisanal fisheries practices in Khong district, Champasack province Lao PDR as a case study. The research focuses on integrating indigenous and scientific knowledge in fisheries management; how local communities use indigenous knowledge to access and manage their fish conservation zones; and the contribution of scientific knowledge to fishery co-management practices at village level. Specific attention is paid to how GIS can aid the integration of these two knowledge systems into a sustainable management system for fisheries resources. Fieldwork was conducted in three villages in the Khong district, Champasack province and Catch per Unit of Effort / hydro-acoustic data collected by the Living Aquatic Resources Research Centre was used to analyse and look at the differences and/or similarities between indigenous and scientific knowledge which can supplement each other and be used for small scale fisheries management. The results show that GIS has the potential not only for data storage and visualisation, but also as a tool to combine scientific and indigenous knowledge in digital maps. Integrating indigenous knowledge into a GIS framework can strengthen indigenous knowledge, from un processed data to information that scientists and decision-makers can easily access and use as a supplement to scientific knowledge in aquatic resource decision-making and planning across different levels. -
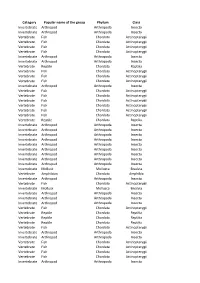
Category Popular Name of the Group Phylum Class Invertebrate
Category Popular name of the group Phylum Class Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Vertebrate Reptile Chordata Reptilia Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Invertebrate Arthropod Arthropoda Insecta Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Fish Chordata Actinopterygii Vertebrate Reptile Chordata Reptilia Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Mollusk Mollusca Bivalvia Vertebrate Amphibian Chordata Amphibia Invertebrate Arthropod Arthropoda Insecta Vertebrate Fish Chordata Actinopterygii Invertebrate Mollusk Mollusca Bivalvia Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Invertebrate Arthropod Arthropoda Insecta Vertebrate -

A Revision of the Species of the Cyprinion Macrostomus-Group (Pisces: Cyprinidae)
ZOBODAT - www.zobodat.at Zoologisch-Botanische Datenbank/Zoological-Botanical Database Digitale Literatur/Digital Literature Zeitschrift/Journal: Annalen des Naturhistorischen Museums in Wien Jahr/Year: 1995 Band/Volume: 97B Autor(en)/Author(s): Herzig-Straschil Barbara, Banarescu Acad. Petru M. Artikel/Article: A revision of the species of the Cyprinion macrostomus-group (Pisces: Cyprinidae). 411-420 ©Naturhistorisches Museum Wien, download unter www.biologiezentrum.at Ann. Naturhist. Mus. Wien 97 B 411 -420 Wien, November 1995 A revision of the species of the Cyprinion macrostomus-group (Pisces: Cyprinidae) P.M. Banarescu* & B. Herzig-Straschil** Abstract Syntypeseries and other material of the five nominal species of the Cyprinion macrostomus-group have been critically examined. On the basis of counts of branched rays in the dorsal fin and number of scales in the lateral line, the shape of the mouth opening and of the dorsal fin, three species are regarded as valid in this group: Cyprinion macrostomus HECKEL, C. kais HECKEL, and C. tenuiradius HECKEL. A lectotype is designated for each of these species. Key words: Cyprinidae, Cyprinion macrostomus-group, revision, lectotype designation. Zusammenfassung Syntypenserien und anderes Material von fünf Arten der Cyprinion macrostomus-Gruppe sind untersucht worden. Auf der Basis der Anzahl der Gabelstrahlen in der Dorsalflosse und der Zahl der Schuppen in der Seitenlinie sowie der Maulform und Ausbildung der Dorsalflosse werden drei Arten in dieser Gruppe aner- kannt: Cyprinion macrostomus HECKEL, C. kais HECKEL und C. tenuiradius HECKEL. Für jede dieser Arten wird ein Lectotypus festgelegt. Introduction Cyprinion HECKEL, 1843 (type species: Cyprinion macrostomus HECKEL, 1843) is a western Asian genus of minnows, distributed from western Syria and the south of the Arabian Peninsula to the western tributaries of Indus River in Punjab (Pakistan). -

Occasional Papers of the Museum of Zoology the University of Michigan
OCCASIONAL PAPERS OF THE MUSEUM OF ZOOLOGY THE UNIVERSITY OF MICHIGAN DISCHERODONTUS, A NEW GENUS OF CYPRINID FISHES FROM SOUTHEASTERN ASIA ABSTRACT.-Rainboth, WalterJohn. 1989. Discherodontus, a new genw of cyprinid firhes from southeastern Asia. Occ. Pap. Mzcs. 2001. Univ. Michigan, 718:I-31, figs. 1-6. Three species of southeast Asian barbins were found to have two rows of pharyngeal teeth, a character unique among barbins. These species also share several other characters which indicate their close relationship, and allow the taxonomic recognition of the genus. Members of this new genus, Discherodontzcs, are found in the Mekong, Chao Phrya, and Meklong basins of Thailand and the Pahang basin of the Malay peninsula. The new genus appears to be most closely related to Chagunizcs of Burma and India, and a group of at least six genera of the southeast Asia-Sunda Shelf basins. Key words: Discherodontus, fihes, Cyprinidae, taxonomy, natural history, Southemt Asia. INTRODUCTION Among the diverse array of barbins of southern and southeastern Asia, there are a number of generic-ranked groups which are poorly understood, or which still await taxonomic recognition. One group of three closely related species, included until now in two genera, is the subject of this paper. Prior to this study, two of the three species in this new genus have been relegated to Puntius of Hamilton (1822), but as understood by Weber and de Beaufort (1916), and by Smith (1945). The remaining species has been placed in Acrossocheilus not of *Department of Biology, University of California (UCLA), Los Angeles, CA 90024 2 Walter J. -

Inventory of Fishes in the Upper Pelus River (Perak River Basin, Perak, Malaysia)
13 4 315 Ikhwanuddin et al ANNOTATED LIST OF SPECIES Check List 13 (4): 315–325 https://doi.org/10.15560/13.4.315 Inventory of fishes in the upper Pelus River (Perak river basin, Perak, Malaysia) Mat Esa Mohd Ikhwanuddin,1 Mohammad Noor Azmai Amal,1 Azizul Aziz,1 Johari Sepet,1 Abdullah Talib,1 Muhammad Faiz Ismail,1 Nor Rohaizah Jamil2 1 Department of Biology, Faculty of Science, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia. 2 Department of Environmental Sciences, Faculty of Environmental Studies, Universiti Putra Malaysia, 43400 UPM Serdang, Selangor, Malaysia. Corresponding author: Mohammad Noor Azmai Amal, [email protected] Abstract The upper Pelus River is located in the remote area of the Kuala Kangsar district, Perak, Malaysia. Recently, the forest along the upper portion of the Pelus River has come under threat due to extensive lumbering and land clearing for plantations. Sampling at 3 localities in the upper Pelus River at 457, 156 and 89 m above mean sea level yielded 521 specimens representing 4 orders, 11 families, 23 genera and 26 species. The most abundant species was Neolissochilus hexagonolepis, followed by Homalopteroides tweediei and Glyptothorax major. The fish community structure indices was observed to increase from the upper to lower portion of the river, which might reflect differences in water velocity. Key words Faunal inventory; freshwater; species diversity; tropical forest. Academinc editor: Barbára Calegari | Received 28 July 2015 | Accepted 27 June 2017 | Published 18 August 2017 Citation: Ikhwanuddin MEM, Amal MNA, Aziz A, Sepet J, Talib A, Ismail MS, Jamil NR (2017). Inventory of fishes in the upper Pelus River (Perak river basin, Perak, Malaysia). -

Viet Nam Ramsar Information Sheet Published on 16 October 2018
RIS for Site no. 2360, Van Long Wetland Nature Reserve, Viet Nam Ramsar Information Sheet Published on 16 October 2018 Viet Nam Van Long Wetland Nature Reserve Designation date 10 February 2017 Site number 2360 Coordinates 20°23'35"N 105°51'10"E Area 2 736,00 ha https://rsis.ramsar.org/ris/2360 Created by RSIS V.1.6 on - 16 October 2018 RIS for Site no. 2360, Van Long Wetland Nature Reserve, Viet Nam Color codes Fields back-shaded in light blue relate to data and information required only for RIS updates. Note that some fields concerning aspects of Part 3, the Ecological Character Description of the RIS (tinted in purple), are not expected to be completed as part of a standard RIS, but are included for completeness so as to provide the requested consistency between the RIS and the format of a ‘full’ Ecological Character Description, as adopted in Resolution X.15 (2008). If a Contracting Party does have information available that is relevant to these fields (for example from a national format Ecological Character Description) it may, if it wishes to, include information in these additional fields. 1 - Summary Summary Van Long Wetland Nature Reserve is a wetland comprised of rivers and a shallow lake with large amounts of submerged vegetation. The wetland area is centred on a block of limestone karst that rises abruptly from the flat coastal plain of the northern Vietnam. It is located within the Gia Vien district of Ninh Binh Province. The wetland is one of the rarest intact lowland inland wetlands remaining in the Red River Delta, Vietnam. -

Chapter I: Introduction 1.1 General Introduction the Mekong River, Along with Its Fourteen Tributaries, Is an Essential Component of the Rural Community in Lao PDR
Chapter I: Introduction 1.1 General introduction The Mekong River, along with its fourteen tributaries, is an essential component of the rural community in Lao PDR. It provides an important source of subsistence for the region, through the supply of animal protein for consumption, such as fish, shrimp, crabs, snails and other aquatic animals. Through fishing, the river provides essential employment and additional household income, as well as supplying water for transportation and agriculture. The decline in fish production in the Mekong River Basin is of increasing concern, not only at the national, but also regional and international levels. Rapid population growth and environmental changes due to the impact of human activity are most likely the main causes of decline in fish numbers (SEAFDEC 2001). Various studies have been undertaken to investigate these issues and to look for alternative solutions to sustainable use of aquatic resources. Fish biology data (fish migration, Catch Per Unit of Effort, hydro-acoustic surveys) and social data (fish production, fish consumption, fish marketing) alone are inadequate for planning and development of natural resources. Indigenous knowledge, however, has shown effective ways to manage fisheries resources in local communities. Differences are evident between the use of scientific based and local knowledge systems. Scientific knowledge is utilised by scientists and is used for decision making at a national level for planning and development. On the other hand, local communities have developed their own management systems based on observation and direct contact with the environment for sustainable development of their resources. However, indigenous knowledge alone is inadequate to deal with fish stock management and planning. -

Biodiversity Assessment of the Mekong River in Northern Lao PDR: a Follow up Study
���� ������������������ ������������������ Biodiversity Assessment of the Mekong River in Northern Lao PDR: A Follow Up Study October, 2004 WANI/REPORT - MWBP.L.W.2.10.05 Follow-Up Survey for Biodiversity Assessment of the Mekong River in Northern Lao PDR Edited by Pierre Dubeau October 2004 The World Conservation Union (IUCN), Water and Nature Initiative and Mekong Wetlands Biodiversity Conservation Programme Report Citation: Author: ed. Dubeau, P. (October 2004) Follow-up Survey for Biodiversity Assessment of the Mekong River in Northern Lao PDR, IUCN Water and Nature Initiative and Mekong Wetlands Biodiversity Conservation and Sustainable Use Programme, Bangkok. i The designation of geographical entities in the book, and the presentation of the material, do not imply the expression of any opinion whatsoever on the part of the Mekong Wetlands Biodiversity Conservation and Sustainable Use Programme (or other participating organisations, e.g. the Governments of Cambodia, Lao PDR, Thailand and Viet Nam, United Nations Development Programme (UNDP), The World Conservation Union (IUCN) and Mekong River Commission) concerning the legal status of any country, territory, or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. The views expressed in this publication do not necessarily reflect those of the Mekong Wetlands Biodiversity Programme (or other participating organisations, e.g. the Governments of Cambodia, Lao PDR, Thailand and Viet Nam, UNDP, The World Conservation Union (IUCN) and Mekong River -

Minnows and Molecules: Resolving the Broad and Fine-Scale Evolutionary Patterns of Cypriniformes
Minnows and molecules: resolving the broad and fine-scale evolutionary patterns of Cypriniformes by Carla Cristina Stout A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama May 7, 2017 Keywords: fish, phylogenomics, population genetics, Leuciscidae, sequence capture Approved by Jonathan W. Armbruster, Chair, Professor of Biological Sciences and Curator of Fishes Jason E. Bond, Professor and Department Chair of Biological Sciences Scott R. Santos, Professor of Biological Sciences Eric Peatman, Associate Professor of Fisheries, Aquaculture, and Aquatic Sciences Abstract Cypriniformes (minnows, carps, loaches, and suckers) is the largest group of freshwater fishes in the world. Despite much attention, previous attempts to elucidate relationships using molecular and morphological characters have been incongruent. The goal of this dissertation is to provide robust support for relationships at various taxonomic levels within Cypriniformes. For the entire order, an anchored hybrid enrichment approach was used to resolve relationships. This resulted in a phylogeny that is largely congruent with previous multilocus phylogenies, but has much stronger support. For members of Leuciscidae, the relationships established using anchored hybrid enrichment were used to estimate divergence times in an attempt to make inferences about their biogeographic history. The predominant lineage of the leuciscids in North America were determined to have entered North America through Beringia ~37 million years ago while the ancestor of the Golden Shiner (Notemigonus crysoleucas) entered ~20–6 million years ago, likely from Europe. Within Leuciscidae, the shiner clade represents genera with much historical taxonomic turbidity. Targeted sequence capture was used to establish relationships in order to inform taxonomic revisions for the clade. -

Native Fish of Conservation Concern in Hong Kong (Part 1)
NATIVE FISH OF CONSERVATION CONCERN IN HONG KONG (PART 1) February 2019 Native Fish of Conservation Concern in Hong Kong (Part 1) Native Fish of Conservation Concern in Hong Kong (Part 1) February 2019 Authors CHENG Hung Tsun & Tony NIP Editors Tony NIP & Gary ADES Contents Contents .......................................................................................................................................... 2 1. Background and Introduction .................................................................................................. 3 2. Methodology ........................................................................................................................... 3 3. Results .................................................................................................................................... 4 4. References .............................................................................................................................. 4 Figure 1. A Hong Kong map with 1 km2 grid lines. ......................................................................... 6 Appendix 1: Anguilla japonica ....................................................................................................... 7 Appendix 2: Anguilla marmorata .................................................................................................. 12 Appendix 3: Acrossocheilus beijiangensis ..................................................................................... 17 Appendix 4: Acrossocheilus parallens ......................................................................................... -

Comparative Phylogeography of Two Codistributed Endemic Cyprinids in Southeastern Taiwan
Biochemical Systematics and Ecology 70 (2017) 283e290 Contents lists available at ScienceDirect Biochemical Systematics and Ecology journal homepage: www.elsevier.com/locate/biochemsyseco Comparative phylogeography of two codistributed endemic cyprinids in southeastern Taiwan Tzen-Yuh Chiang a, 1, Yi-Yen Chen a, 1, Teh-Wang Lee b, Kui-Ching Hsu c, * Feng-Jiau Lin d, Wei-Kuang Wang e, Hung-Du Lin f, a Department of Life Sciences, Cheng Kung University, Tainan 701, Taiwan b Taiwan Endemic Species Research Institute, ChiChi, Nantou 552, Taiwan c Department of Industrial Management, National Taiwan University of Science and Technology, Taipei 10607, Taiwan d Tainan Hydraulics Laboratory, National Cheng Kung University, Tainan 709, Taiwan e Department of Environmental Engineering and Science, Feng Chia University, Taichung 407, Taiwan f The Affiliated School of National Tainan First Senior High School, Tainan 701, Taiwan article info abstract Article history: Two cyprinid fishes, Spinibarbus hollandi and Onychostoma alticorpus, are endemic to Received 12 October 2016 southeastern Taiwan. This study examined the phylogeography of these two codistributed Received in revised form 3 December 2016 primary freshwater fishes using mitochondrial DNA cytochrome b sequences (1140 bp) to Accepted 18 December 2016 search for general patterns in the effect of historical changes in southeastern Taiwan. In total, 135 specimens belonging to these two species were collected from five populations. These two codistributed species revealed similar genetic variation patterns. The genetic Keywords: variation in both species was very low, and the geographical distribution of the genetic Spinibarbus hollandi Onychostoma alticorpus variation corresponded neither to the drainage structure nor to the geographical distances Mitochondrial between the samples.