Selective Oxidation of Polynuclear Aromatic Hydrocarbons
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Selective Hydrogenation of Cyclopentadiene to Form Cyclopentene
European Patent Office (n) Publication number: 0 009 035 Office europeen des brevets A1 EUROPEAN PATENT APPLICATION {ry Application number: 79930020.7 © lnt.CI.3: C 07 C 13/12 C 07 C 5/05, B 01 J 31/14 (22) Date of filing: 27.08.79 B 01 J 31/12 (30) Priority: 30.08.78 US 938159 (71) Applicant: THE GOODYEAR TIRE 8t RUBBER COMPANY 1144 East Market Street Akron, Ohio, 44316IUS) i*3) uaie ot puDiication ot application: 19.03.80 Bulletin 80 6 [73) Inventor: Menapace, Henry Robert 1738 Washington Circle @ Designated Contracting States: Stow Ohio 44224(US) BE DE FR GB IT NL g) Representative: Zewen, Arthur 4 Place Winston Churchill B.P. 447 Luxembourg-VillelLU ) {&) Selective hydrogenation of cyclopentadiene to form cyclopentene. where (57) Theremere is oiscloseddisclosed a process torfor the preparation otof cyc- wnere Hi,R1, H2R2 and R3 may bebe hydrogen or halogen, or an alkyl lopentene which comprises selectively hydrogenating cyc- radical containing from 1 to 6 carbon atoms; R,OR,R1OR2 wherein lopentadiene in a liquid phase by contacting cyclopentadiene R,Ri and R2 may bebe the same or different alkyl radicals contain- from with hydrogen in the presence of a hydrogenation catalyst inging 1 to 6 carbon atoms; and R^CR^ comprising (1(1) ) a soluble nickel compound, (2) an 0wherein wherein R,R1 may be organoaluminum compound or a lithium alkyl compound, alkylilkyl or an aromatic radical containing from 1 to 8 and (3) at least selected from the carbon one cocatalyst compound atomsitoms and RsR2 may be hydrogen or an alkyl or aromatic hyd- group consisting of HjO;H2O; NH3; ROH where R is an alkyl or a rocarbonocarbon radical containing from 1 to 8 carbon atoms. -

Cyclopentyl Methyl Ether (CPME)
Korean J. Chem. Eng., 34(2), 463-469 (2017) pISSN: 0256-1115 DOI: 10.1007/s11814-016-0265-5 eISSN: 1975-7220 INVITED REVIEW PAPER Measurement and correlation of the isothermal VLE data for the binary mixtures of cyclopentene (CPEN)+cyclopentyl methyl ether (CPME) Wan Ju Jeong and Jong Sung Lim† Department of Chemical and Biomolecular Engineering, Sogang University, C.P.O. Box 1142, Seoul 04107, Korea (Received 2 August 2016 • accepted 20 September 2016) Abstract−The isothermal vapor-liquid equilibrium data for the binary systems of cyclopentene (1)+cyclopentyl methyl ether (2) were measured at 313.15, 323.15, 333.15, 343.15 and 353.15 K using a dynamic-type equilibrium apparatus and online gas chromatography analysis. For all the measured VLE data consistency tests were performed for the verifi- cation of data using Barker’s method and the ASPEN PLUS Area Test method. All the resulting average absolute val- δ γ γ ues of residuals [ ln ( 1/ 2)] for Barker’s method and D values for the ASPEN PLUS area test method were com- paratively small. So, the VLE data reported in this study are considered to be acceptable. This binary system shows neg- ative deviation from Raoult’s law and does not exhibit azeotropic behavior at whole temperature ranges studied here. The measured data were correlated with the P-R EoS using the Wong-Sandler mixing rule. The overall average relative deviation of pressure (ARD-P (%)) between experimental and calculated values was 0.078% and that of vapor phase compositions (ARD-y (%)) was 0.452%. Keywords: Vapor Liquid Equilibria (VLE), Cyclopentyl Methyl Ether (CPME), Cyclopentene (CPEN), Peng-Robinson Equation of State (PR-EoS), Wong-Sandler Mixing Rule (WS-MR) INTRODUCTION however, have some disadvantages because they use dimethyl sul- fate and methyl iodide as a reactant, respectively, which are muta- Ethers are widely used in organic chemistry and biochemistry. -

Secondary Organic Aerosol Formation from the Ozonolysis of Cycloalkenes and Related Compounds*
252 Chapter 7 Secondary Organic Aerosol Formation from the Ozonolysis of Cycloalkenes and Related Compounds* * This chapter is reproduced by permission from “Secondary organic aerosol formation from ozonolysis of cycloalkenes and related compounds” by M.D. Keywood, V. Varutbangkul, R. Bahreini, R.C. Flagan, J.H. Seinfeld, Environmental Science and Technology, 38 (15): 4157-4164, 2004. Copyright 2004, American Chemical Society. 253 7.1. Abstract The secondary organic aerosol (SOA) yields from the laboratory chamber ozonolysis of a series of cycloalkenes and related compounds are reported. The aim of this work is to investigate the effect of the structure of the hydrocarbon parent molecule on SOA formation for a homologous set of compounds. Aspects of the compound structures that are varied include the number of carbon atoms present in the cycloalkene ring (C5 to C8), the presence and location of methyl groups, and the presence of an exocyclic or endocyclic double bond. The specific compounds considered here are cyclopentene, cyclohexene, cycloheptene, cyclooctene, 1-methyl-1-cyclopentene, 1-methyl-1- cyclohexene, 1-methyl-1-cycloheptene, 3-methyl-1-cyclohexene, and methylene cyclohexane. SOA yield is found to be a function of the number of carbons present in the cycloalkene ring, with increasing number resulting in increased yield. Yield is enhanced by the presence of a methyl group located at a double-bonded site but reduced by the presence of a methyl group at a non-double bonded site. The presence of an exocyclic double bond also leads to a reduced yield relative to the equivalent methylated cycloalkene. On the basis of these observations, the SOA yield for terpinolene relative to the other cyclic alkenes is qualitatively predicted, and this prediction compares well to measurements of SOA yield from the ozonolysis of terpinolene. -

(12) United States Patent (10) Patent No.: US 7.494,962 B2 Kinet Al
USOO74949.62B2 (12) United States Patent (10) Patent No.: US 7.494,962 B2 Kinet al. (45) Date of Patent: Feb. 24, 2009 (54) SOLVENTS CONTAINING CYCLOAKYL (56) References Cited ALKYLETHERS AND PROCESS FOR PRODUCTION OF THE ETHERS U.S. PATENT DOCUMENTS 3496,223 A * 2/1970 Mitchell et al. ............... 562/22 (75) Inventors: Idan Kin, Ottawa (CA); Genichi Ohta, Tokyo (JP); Kazuo Teraishi, Tokyo (JP); Kiyoshi Watanabe, Tokyo (JP) (Continued) (73) Assignee: Zeon Corporation, Tokyo (JP) FOREIGN PATENT DOCUMENTS (*) Notice: Subject to any disclaimer, the term of this EP 587434 A1 3, 1994 patent is extended or adjusted under 35 U.S.C. 154(b) by 349 days. (Continued) (21) Appl. No.: 10/481,340 OTHER PUBLICATIONS (22) PCT Filed: Jun. 27, 2002 Edited by Kagaku Daijiten Henshu Iinkai, “Kagaku Daijiten 9”. (86). PCT No.: PCT/UP02/06501 Kyoritsu Shuppan Co., Ltd., Aug. 25, 1962, p. 437, "Yozai'. (Continued) S371 (c)(1), (2), (4) Date: Sep. 24, 2004 Primary Examiner Gregory E Webb (74) Attorney, Agent, or Firm—Birch, Stewart, Kolasch & (87) PCT Pub. No.: WO03/002500 Birch, LLP PCT Pub. Date: Jan. 9, 2003 (57) ABSTRACT (65) Prior Publication Data The present inventions are (A) a solvent comprising at least US 2005/OO65060A1 Mar. 24, 2005 one cycloalkyl alkyl ether (1) represented by the general O O formula: R1-O R2 (wherein R1 is cyclopentyl or the like: (30) Foreign Application Priority Data and R2 is C1-10 alkyl or the like); (B) a method of prepara Jun. 28, 2001 (JP) ............................. 2001-196766 tions the ethers (1) characterized by reacting an alicyclic Oct. -

Alfa Olefins Cas N
OECD SIDS ALFA OLEFINS FOREWORD INTRODUCTION ALFA OLEFINS CAS N°:592-41-6, 111-66-0, 872-05-9, 112-41-4, 1120-36-1 UNEP PUBLICATIONS 1 OECD SIDS ALFA OLEFINS SIDS Initial Assessment Report For 11th SIAM (Orlando, Florida, United States 1/01) Chemical Name: 1-hexene Chemical Name: 1-octene CAS No.: 592-41-6 CAS No.: 111-66-0 Chemical Name: 1-decene Chemical Name: 1-dodecene CAS No.: 872-05-9 CAS No.: 112-41-4 Chemical Name: 1-tetradecene CAS No.: 1120-36-1 Sponsor Country: United States National SIDS Contract Point in Sponsor Country: United States: Dr. Oscar Hernandez Environmental Protection Agency OPPT/RAD (7403) 401 M Street, S.W. Washington, DC 20460 Sponsor Country: Finland (for 1-decene) National SIDS Contact Point in Sponsor Country: Ms. Jaana Heiskanen Finnish Environment Agency Chemicals Division P.O. Box 140 00251 Helsinki HISTORY: SIDS Dossier and Testing Plan were reviewed at the SIDS Review Meeting or in SIDS Review Process on October 1993. The following SIDS Testing Plan was agreed: No testing ( ) Testing (x) Combined reproductive/developmental on 1-hexene, combined repeat dose/reproductive/developmental on 1-tetradecene and acute fish, daphnid and algae on 1- tetradecene. COMMENTS: The following comments were made at SIAM 6 and have been incorporated in this version of the SIAR: 2 UNEP PUBLICATIONS OECD SIDS ALFA OLEFINS 1. The use of QSAR calculations for aquatic toxicity, 2. More quantitative assessment of effects; and 3. Provide more details for each endpoint. The following comments were made at SIAM 6, but were not incorporated into the SIAR for the reasons provided: 1. -

Radical Approaches to Alangium and Mitragyna Alkaloids
Radical Approaches to Alangium and Mitragyna Alkaloids A Thesis Submitted for a PhD University of York Department of Chemistry 2010 Matthew James Palframan Abstract The work presented in this thesis has focused on the development of novel and concise syntheses of Alangium and Mitragyna alkaloids, and especial approaches towards (±)-protoemetinol (a), which is a key precursor of a range of Alangium alkaloids such as psychotrine (b) and deoxytubulosine (c). The approaches include the use of a key radical cyclisation to form the tri-cyclic core. O O O N N N O O O H H H H H H O N NH N Protoemetinol OH HO a Psychotrine Deoxytubulosine b c Chapter 1 gives a general overview of radical chemistry and it focuses on the application of radical intermolecular and intramolecular reactions in synthesis. Consideration is given to the mediator of radical reactions from the classic organotin reagents, to more recently developed alternative hydrides. An overview of previous synthetic approaches to a range of Alangium and Mitragyna alkaloids is then explored. Chapter 2 follows on from previous work within our group, involving the use of phosphorus hydride radical addition reactions, to alkenes or dienes, followed by a subsequent Horner-Wadsworth-Emmons reaction. It was expected that the tri-cyclic core of the Alangium alkaloids could be prepared by cyclisation of a 1,7-diene, using a phosphorus hydride to afford the phosphonate or phosphonothioate, however this approach was unsuccessful and it highlighted some limitations of the methodology. Chapter 3 explores the radical and ionic chemistry of a range of silanes. -

Epoxidation of Cyclopentene, Cyclohexene and Cycloheptene by Acetylperoxyl Radicals
Epoxidation of Cyclopentene, Cyclohexene and Cycloheptene by Acetylperoxyl Radicals J. R. Lindsay Smith, D. M. S. Smith, M. S. Stark* and D. J. Waddington Department of Chemistry, University of York, York, YO10 5DD, United Kingdom To further the current understanding of how peroxyl radicals add to C=C double bonds,eg. 1-3 a series of cyclic alkenes (cyclopentene, cyclohexene and cycloheptene) were reacted with acetylperoxyl radicals over the temperature range 373 - 433 K, via the co- oxidation of acetaldehyde, the cyclic alkene and a reference alkene. The resulting epoxides were monitored by GC-FID, allowing the determination of the Arrhenius parameters given in Table 1 for these addition reactions. 3 -1 -1 -1 Alkene log10(A / dm mol s ) Eact / kJ mol cyclopentene 9.7±0.6 31.2±5.0 cyclohexene 7.7±0.7 17.4±5.3 cycloheptene 8.8±0.8 25.5±6.5 cis-2-butene4 8.1±0.5 22.9±3.8 Table 1: Arrhenius parameters for the addition of acetylperoxyl radicals to cyclic alkenes and cis-2-butene. Parameters for cis-2-butene are also given for comparison,4 as it would be expected that a relatively unstrained cycloalkene would behave in a similar manner to this non-cyclic alkene; this is indeed found to be so, with no statistically significant difference between cyclohexene and cis-2-butene. Indeed the measured activation energy for cyclohexene fits in well with correlation between charge transfer ( Ec) and activation energy that is known to exist for the addition of acetylperoxyl to acyclic mono-alkenes.1 Cyclopentene however does have a significantly larger pre-exponential factor and activation energy than either cyclohexene or cis-2-butene. -

A Comparative Study of the Formation of Aromatics In
A COMPARATIVE STUDY OF THE FORMATION OF AROMATICS IN RICH METHANE FLAMES DOPED BY UNSATURATED COMPOUNDS H.A. GUENICHE, J. BIET, P.A. GLAUDE, R. FOURNET, F. BATTIN-LECLERC* Département de Chimie-Physique des Réactions, Nancy Université, CNRS, 1 rue Grandville, BP 20451, 54001 NANCY Cedex, France * E-mail : [email protected] ; Tel.: 33 3 83 17 51 25 , Fax : 33 3 83 37 81 20 ABSTRACT For a better modeling of the importance of the different channels leading to the first aromatic ring, we have compared the structures of laminar rich premixed methane flames doped with several unsaturated hydrocarbons: allene and propyne, because they are precursors of propargyl radicals which are well known as having an important role in forming benzene, 1,3-butadiene to put in evidence a possible production of benzene due to reactions of C4 compounds, and, finally, cyclopentene which is a source of cyclopentadienylmethylene radicals which in turn are expected to easily isomerizes to give benzene. These flames have been stabilized on a burner at a pressure of 6.7 kPa (50 Torr) using argon as dilutant, for equivalence ratios (φ) from 1.55 to 1.79. A unique mechanism, including the formation and decomposition of benzene and toluene, has been used to model the oxidation of allene, propyne, 1,3-butadiene and cyclopentene. The main reaction pathways of aromatics formation have been derived from reaction rate and sensitivity analyses and have been compared for the three types of additives. These combined analyses and comparisons can only been performed when a unique mechanism is available for all the studied additives. -

The Arene–Alkene Photocycloaddition
The arene–alkene photocycloaddition Ursula Streit and Christian G. Bochet* Review Open Access Address: Beilstein J. Org. Chem. 2011, 7, 525–542. Department of Chemistry, University of Fribourg, Chemin du Musée 9, doi:10.3762/bjoc.7.61 CH-1700 Fribourg, Switzerland Received: 07 January 2011 Email: Accepted: 23 March 2011 Ursula Streit - [email protected]; Christian G. Bochet* - Published: 28 April 2011 [email protected] This article is part of the Thematic Series "Photocycloadditions and * Corresponding author photorearrangements". Keywords: Guest Editor: A. G. Griesbeck benzene derivatives; cycloadditions; Diels–Alder; photochemistry © 2011 Streit and Bochet; licensee Beilstein-Institut. License and terms: see end of document. Abstract In the presence of an alkene, three different modes of photocycloaddition with benzene derivatives can occur; the [2 + 2] or ortho, the [3 + 2] or meta, and the [4 + 2] or para photocycloaddition. This short review aims to demonstrate the synthetic power of these photocycloadditions. Introduction Photocycloadditions occur in a variety of modes [1]. The best In the presence of an alkene, three different modes of photo- known representatives are undoubtedly the [2 + 2] photocyclo- cycloaddition with benzene derivatives can occur, viz. the addition, forming either cyclobutanes or four-membered hetero- [2 + 2] or ortho, the [3 + 2] or meta, and the [4 + 2] or para cycles (as in the Paternò–Büchi reaction), whilst excited-state photocycloaddition (Scheme 2). The descriptors ortho, meta [4 + 4] cycloadditions can also occur to afford cyclooctadiene and para only indicate the connectivity to the aromatic ring, and compounds. On the other hand, the well-known thermal [4 + 2] do not have any implication with regard to the reaction mecha- cycloaddition (Diels–Alder reaction) is only very rarely nism. -
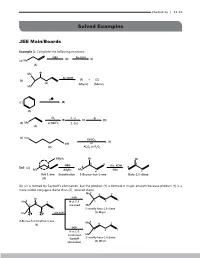
11.Alkenes-And-Alkynesexercise.Pdf
Chemistry | 11.43 Solved Examples JEE Main/Boards Example 1: Complete the following reactions: NBS Alc.KOH (B) (C ) (a) Me (A) Me Br Alc.KOH (b) (Y) + (Z) (X) (Major) (Minor) Me H (c ) (B) (A) Cl 1. Li -B 2 (B) (C ) (D) (d) Me at 500oC 2. CuI (A) (e) HO KHSO 4 (Y) OH or (X) Al23O or PO25 Allylic Br Br 3 1 3 NBS Alc. KOH Sol: (a) 4 4 Me Allylic Me 2 -Hbr 2 But-1-ene Substitution 3-Bromo-but-1-ene Buta-1,3-diene (A) (b) (Z) is formed by Saytzeff’s elimination, but the product (Y) is formed in major amount because product (Y) is a more stable conjugate diene than (Z), isolated diene 6 Me 4 2 -HBr Br 1 Me6 H at C-3 5 3 4 2 removed Me (b) 5 3 1 5-methly hexa-1,3-diene Me H H Alk.KOH (Y) Major 4-Bromo-5-methylhex-1-ene Me6 (X) 4 2 -HBr 5 1 H at C-3 3 is removed Me Saytzeff 5-methly hexa-1,4-diene elimination (Z) Minor 11.44 | Alkenes and Alkynes (c) 4 1 H H 3 5 -H 2 6 ( c ) H H 2 6 3 5 1 4 Less stable More stable isolated diene conjugated diene (A) (B) Cyclohexa-1,4-diene Cyclohexa-1,3-diene 2 o 2 Cl at 500 2 4 2 3 1. Li (d) 1 1 1 Me Allylic 2. Cu 3 (d) Prop-1-ene substitution Cl Corey-House Allylic synthesis Diallyl R (A) 3-Chloro- lithium cuperate prop-1-ene (B) Cl 4 4 6 1 3 5 R’ H (e) (e) HO 2 4 KHSO4 OH Or 1 3 H Al23O or PO25 (X) (Y) (-H2O) Butan-1,4-diol Butan-1,3-diene Example 2: Give the major products (not stereoisomers) of the following: 5 5 (a 1 4 (a)(a) 1 4 22 33 Cyclopenta-1,3-diene 1 1mo mol l 1 mo1 mol l 1 mo1l mol 11 mol of BrBr22 HBrHBr + + HBHBr r HS2 +HS + peroxide 2 peroxide peroperoxide xide (b) 55 (b) 1 (b) Me 1 4 Me 4 2 3 3 1-Ethylcyclopenta-1,3-diene2 1-Ethylcyclopenta-1,3-diene 1 mol 1 mol 1 mol 1 mol of Br2 1 mol of Br HBr1 mo + l HBr1 mol 1 mol 2 HS2 + peroHBrxide + HBr peroHSxide2 + peroxide peroxide Chemistry | 11.45 Sol: 1 (a) (a) 2 5 3 4 (A) All 1,4-addition products HBr or HS+ Br2 2 1,4-Add. -

Self-Metathesis of 1-Octene Using Alumina-Supported Re2o7 in Supercritical CO2
Top Catal DOI 10.1007/s11244-008-9154-4 ORIGINAL PAPER Self-Metathesis of 1-Octene Using Alumina-Supported Re2O7 in Supercritical CO2 Massimo Fabris Æ Cindy Aquino Æ Antony J. Ward Æ Alvise Perosa Æ Thomas Maschmeyer Æ Maurizio Selva Ó Springer Science+Business Media, LLC 2009 Abstract In this contribution we describe the use of basic science has been applied for the benefit of man, heterogeneous catalysts for the liquid-phase self-metathesis society and the environment’’.1 Their contribution was of 1-octene in supercritical CO2. Our work aims at recognized for understanding the mechanism, and for addressing the mass-transfer problems associated with such developing very efficient and selective discrete metal- reaction systems. By coupling a heterogeneous supported based catalysts. Re2O7 catalyst with the use of scCO2, the self-metathesis Nonetheless, one should not forget that the metathesis of 1-octene takes place by and large much more rapidly reaction using heterogeneous catalysis has been an estab- than in traditional solvent media, and furthermore, by using lished industrial process since 1966, based on the use of scCO2 the overall efficiency and sustainability of the supported (usually on silica and alumina) metal oxides transformation can be improved. such as tungsten, nickel, cobalt, rhenium, and molybde- num. Three significant examples come to mind and are Keywords Metathesis Á 1-Octene Á Supercritical CO2 Á here recalled briefly. [1]. Re/Al O 2 3 1. The historic Phillips triolefin process for the produc- tion of ethene and 2-butene from propene using a WO /SiO catalyst, [2] that was shut down in 1972 1 Introduction 3 2 after 6 years of operation due to increased demand for propene, and later reutilized in the reverse direction to The metathesis of olefins is a very elegant and widely produce propene. -

Desymmetrisation Reactions of Cyclohexa-1,4- Dienes and Marine Natural Product Synthesis Joe Hill-Cousins UMI Number: U585B54
Desymmetrisation reactions of cyclohexa-1,4- dienes and marine natural product synthesis Joe Hill-Cousins UMI Number: U585B54 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. Dissertation Publishing UMI U585354 Published by ProQuest LLC 2013. Copyright in the Dissertation held by the Author. Microform Edition © ProQuest LLC. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 Abstract This thesis describes novel diastereoselective group selective processes for the desymmetrisation of cyclohexa-1,4-dienes, and their application to target synthesis of the cladiellin diterpenes. In addition, an enantioselective approach to batzelladine C methyl ester is described, permitting stereochemical assignment of batzelladine C. Chapter 1 describes the use of diastereoselective iodocyclisation reactions for the desymmetrisation of cyclohexa-1,4-dienes, and also details a novel epoxidation-cyclisation transformation, enabling the formation of up to six contiguous stereogenic centres, selectively, in a single step. Chapter 2 introduces the cladiellin diterpenes and describes the application of novel diastereoselective Prins chemistry for the synthesis of a model of the cladiellin core. Chapter 3 describes our attempts at elaborating the cladiellin model to approach the cladiellin framework and discusses an unexpected but interesting Prins cyclisation/rearrangement process.