Controlled Potential Coulometry in Non-Aqueous Solvents
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
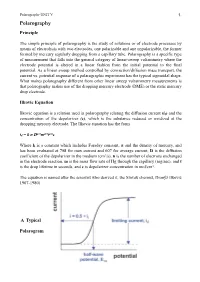
Polarography.Pdf
Polarography UNIT V !1. Polarography Principle The simple principle of polarography is the study of solutions or of electrode processes by means of electrolysis with two electrodes, one polarizable and one unpolarizable, the former formed by mercury regularly dropping from a capillary tube. Polarography is a specific type of measurement that falls into the general category of linear-sweep voltammetry where the electrode potential is altered in a linear fashion from the initial potential to the final potential. As a linear sweep method controlled by convection/diffusion mass transport, the current vs. potential response of a polarographic experiment has the typical sigmoidal shape. What makes polarography different from other linear sweep voltammetry measurements is that polarography makes use of the dropping mercury electrode (DME) or the static mercury drop electrode. Ilkovic Equation Ilkovic equation is a relation used in polarography relating the diffusion current (id) and the concentration of the depolarizer (c), which is the substance reduced or oxidized at the dropping mercury electrode. The Ilkovic equation has the form id = k n D1/3m2/3t1/6c Where k is a constant which includes Faraday constant, π and the density of mercury, and has been evaluated at 708 for max current and 607 for average current, D is the diffusion coefficient of the depolarizer in the medium (cm2/s), n is the number of electrons exchanged in the electrode reaction, m is the mass flow rate of Hg through the capillary (mg/sec), and t is the drop lifetime in seconds, and c is depolarizer concentration in mol/cm3. The equation is named after the scientist who derived it, the Slovak chemist, Dionýz Ilkovič 1907-1980). -

07 Chapter2.Pdf
22 METHODOLOGY 2.1 INTRODUCTION TO ELECTROCHEMICAL TECHNIQUES Electrochemical techniques of analysis involve the measurement of voltage or current. Such methods are concerned with the interplay between solution/electrode interfaces. The methods involve the changes of current, potential and charge as a function of chemical reactions. One or more of the four parameters i.e. potential, current, charge and time can be measured in these techniques and by plotting the graphs of these different parameters in various ways, one can get the desired information. Sensitivity, short analysis time, wide range of temperature, simplicity, use of many solvents are some of the advantages of these methods over the others which makes them useful in kinetic and thermodynamic studies1-3. In general, three electrodes viz., working electrode, the reference electrode, and the counter or auxiliary electrode are used for the measurement in electrochemical techniques. Depending on the combinations of parameters and types of electrodes there are various electrochemical techniques. These include potentiometry, polarography, voltammetry, cyclic voltammetry, chronopotentiometry, linear sweep techniques, amperometry, pulsed techniques etc. These techniques are mainly classified into static and dynamic methods. Static methods are those in which no current passes through the electrode-solution interface and the concentration of analyte species remains constant as in potentiometry. In dynamic methods, a current flows across the electrode-solution interface and the concentration of species changes such as in voltammetry and coulometry4. 2.2 VOLTAMMETRY The field of voltammetry was developed from polarography, which was invented by the Czechoslovakian Chemist Jaroslav Heyrovsky in the early 1920s5. Voltammetry is an electrochemical technique of analysis which includes the measurement of current as a function of applied potential under the conditions that promote polarization of working electrode6. -

Unit 1 Introduction to Electro- Analytical Methods
Introduction to UNIT 1 INTRODUCTION TO ELECTRO- Electroanalytical ANALYTICAL METHODS Methods Structure 1.1 Introduction Objectives 1.2 Basic Concepts Electrical Units Basic Laws of Electrochemistry Electrode Potential Liquid-Junction Potentials Electrochemical Cells The Nernst Equation Cell Potential 1.3 Classification and an Overview of Electroanalytical Methods Potentiometry Voltammetry Polarography Amperometry Electrogravimetry and Coulometry Conductometry 1.4 Classification and Relationships of Electroanalytical Methods 1.5 Summary 1.6 Terminal Questions 1.7 Answers 1.1 INTRODUCTION This is the first unit of this course. This unit deals with the fundamentals of electrochemistry that are necessary for understanding the principles of electroanalytical methods discussed in this Unit 2 to 9. In this unit we have also classified of electroanalytical methods and briefly introduced of some important electroanalytical methods. More details of these elecroanalytical methods will be discussed in the consecutive units. Objectives After studying this unit, you will be able to: • name the different units of electrical quantities, • define the two basic laws of electrochemistry, • describe the single electrode potential and the potential of a galvanic cell, • derive the Nernst expression and give its applications, • calculate the electrode potentials and cell potentials using Nernst equation, • describe the basis for classification of the electroanalytical techniques, and • explain the basis principles and describe the essential conditions of the various electroanalytical techniques. 1.2 BASIC CONCEPTS Before going in detail of different electroanalytical techniques, let’s recapitulate some basic concepts which you have studied in your undergraduate classes. 7 Electroanalytical 1.2.1 Electrical Units Methods -I Ampere (A): Ampere is the unit of current. -

Lecture Content
EMT 518: METHODS IN ENVIROMENTAL ANALYSIS III (2 UNITS) Lecturer: Professor O. Bamgbose SYNOPSIS Electro-analytical method: Potentiometry, Reference electrode – Calomel, Ag/Agcl, indicator electrodes – 1st, 2nd and 3rd order, Metal Electrodes, membrane electrodes – glass electrode, types of liquid junction potential, solid state electrode, potentiometric titration, end point location in potentiometric titration –visual, plot of E/V, plot of derivative curves 1st and 2nd electrogravimetry, fixed potential, constant current, constant cathode potential coulometry: constant current coulometry, coulometric titration. Voltammetry: classical polarography, Description of dropping mercury electrode, condition for polarographic determination, qualitative and quantitative analysis conductance methods: description of limiting ionic conductance, conductance cell, conductomertic titration. Thermal methods: Thermogravimetry, differential thermal analysis (DTA) LECTURE CONTENT POTENTIOMETRY Is a measurement of a given chemical species in an equilibrium system by the use of an electrode, while potentiometric titration is the technique that is used for following the changes in the concentration of chemical species as function of added titrant using an electrode. In both cases a cell is needed and a cell consists of the following: (1) Reference electrode (2) Liquid junction (3) Analyte solution (4) indicator electrode. It is also possible to have a cell without liquid junction. REFERENCE ELECTRODE. In carrying out a potentiometric determination the half cell potential of one electrode must be known which should be constant, reproducible and completely insensitive to the reference electrode and must be fully polarised throughout the duration of the measurement i.e the potential of the reference electrode does not change through the whole measurement. A classical example of reference electrode is the calomel electrode. -

Physical Electrochemical Software Brochure
Redefining Electrochemical Measurement Physical Electrochemistry Software The Physical Electrochemistry Software is used with a to the limit. You can define the potentials as absolute Gamry Potentiostat to perform in-depth studies of the voltages or by their relationship to the Open-Circuit structure of the electrode interface and the mechanisms Potential. of electrochemical reactions. The software brings Cyclic Voltammetry and other recognized electrochemical The scan rate (mV/s) is determined by the interval between research techniques to the Gamry user. The Physical data points (sample period) and the Step Size (mV): Electrochemistry Software is a useful tool for Step Size() mV fundamental studies, sensor development, small-scale Scan Rate() mV s = energy storage devices, electrophysiology, etc. Sample Period() s The minimum sample period may be as low as 3.3 µs. The This software incorporates the following electrochemical maximum Scan Rate is a function of Step Size. For techniques: example, the maximum Scan Rate with a 2 mV step is 600 V/s. Higher steps provide faster scan rates, but at the ••• Cyclic Voltammetry expense of resolution. Step Sizes greater than 10 mV are ••• Linear Sweep Voltammetry likely to result in unsatisfactory data. ••• Chronoamperometry ••• Repeating Chronoamperometry The Physical Electrochemistry Software can save, and ••• MMuullttiipplleeMultiple-Multiple---StepStep Chronoamperometry display, up to 262,143 data points! The number of CV ••• ChronopotentChronopotentiometryiioommeettrryyiometry cycles that can be displayed is dependent upon the scan ••• Repeating Chronopotentiometry parameters. ••• Chronocoulometry Step Size() mV No. of Cycles =262,143 × ••• Controlled Potential Coulometry Voltage Span of theCV() mV Like most Gamry software, the Physical Electrochemistry Software and a Gamry Potentiostat use the Framework for data acquisition and the Echem Analyst for data analysis. -

Stationary Electrode Voltammetry and Chronoamperometry in an Alkali Metal Carbonate-Borate Melt
AN ABSTRACT OF THE THESIS OF DARRELL GEORGE PETCOFF for the Doctor of Philosophy (Name of student) (Degree) in Analytical Chemistry presented onC (O,/97 (Major) (Date) Title: STATIONARY ELECTRODE VOLTAMMETRY AND CHRONOAMPEROMETRY IN AN ALKALI METAL CARBONATE - BORATE. MFT T Abstract approved: Redacted for Privacy- Drir. reund The electrochemistry of the lithium-potassium-sodium carbonate-borate melt was explored by voltammetry and chrono- amperometry. In support of this, a controlled-potential polarograph and associated hardware was constructed.Several different types of reference electrodes were tried before choosing a porcelain mem- brane electrode containing a silver wire immersed in a silver sulfate melt.The special porcelain compounded was used also to construct a planar gold disk electrode.The theory of stationary electrode polarography was summarized and denormalized to provide an over- all view. A new approach to the theory of the cyclic background current was also advanced. A computer program was written to facilitate data processing.In addition to providing peak potentials, currents, and n-values, the program also resolves overlapping peaks and furnishes plots of both processed and unprocessed data. Rapid-scan voltammetry was employed to explore the electro- chemical behavior of Zn, Co, Fe, Tl, Sb, As, Ni, Sn, Cd, Te, Bi, Cr, Pb, Cu, and U in the carbonate-borate melt. Most substances gave reasonably well-defined peaks with characteristic peak potentials and n-values.Metal deposition was commonly accompanied by adsorp- tion prepeaks indicative of strong adsorption, and there was also evi- dence of a preceding chemical reaction for several elements, sug- gesting decomplexation before reduction. -

Thesis-1961-B586i.Pdf
INVESTIGATION OF SOME POSSIBILITIES FOR AMPEROMETRIC TITRATION OF CERTAIN METAL IONS WITH OXINE By Donald George Biechler I I Bachelor of Science University of Wisconsin Madison, Wisconsin 1956 Submitted to the faculty of the Graduate School of the Oklahoma State University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE May, 1961 INVESTIGATION OF SOME POSSIBILITIES FOR AMPEROMEI'RIC TITRATION OF CERTAIN MEI'AL IONS WITH OXINE Thesis Approved: Thesis Adviser i i OKLAHOMA STATE UNIVERSITY llBRARY JAN 2 1962 PREFACE Oxine (8-hydroxyquinoline) is most generally used in analytical chemistry as a precipitant for metals and is known to form water- insoluble chelates with better than thirty metal ions (3). There exists in solutions of oxine a tautomeric equilibrium of the fol- lowing type: C C C C /~/'\ ,c/""/~ C C f ij 1 I II I C C C C C C ~/"'/C N ~/C "+/N r . _I I 0 H 0--------H Chelation of a metal ion involves replacement of the proton and for- mation of a coordinate bond with the nitrogen to form a stable 5 membered ring compound. Thus nickel, a bivalent cation, would form a compound with the following structure: 4810 90 iii iv The oxinates can be ignited and weighed as such or they may be further ignited to the metal oxides and then weighed. Alternately the oxinates may be dissolved in acid and quantitatively brominated (7)0 Considering the number of metal ions that are precipitated by oxine, it seemed that possibly more use could be made of the reagent in volumetric analysis. -

COULOMETRY for the DETERMINATION of URANIUM and PLUTONIUM: PAST and PRESENT by M.K
BARC/2012/E/001 BARC/2012/E/001 COULOMETRY FOR THE DETERMINATION OF URANIUM AND PLUTONIUM: PAST AND PRESENT by M.K. Sharma, J.V. Kamat, A.S. Ambolikar, J.S. Pillai and S.K. Aggarwal Fuel Chemistry Division 2012 BARC/2012/E/001 GOVERNMENT OF INDIA ATOMIC ENERGY COMMISSION BARC/2012/E/001 COULOMETRY FOR THE DETERMINATION OF URANIUM AND PLUTONIUM: PAST AND PRESENT by M.K. Sharma, J.V. Kamat, A.S. Ambolikar, J.S. Pillai and S.K. Aggarwal Fuel Chemistry Division BHABHA ATOMIC RESEARCH CENTRE MUMBAI, INDIA 2012 BARC/2012/E/001 BIBLIOGRAPHIC DESCRIPTION SHEET FOR TECHNICAL REPORT (as per IS : 9400 - 1980) 01 Security classification : Unclassified 02 Distribution : External 03 Report status : New 04 Series : BARC External 05 Report type : Technical Report 06 Report No. : BARC/2012/E/001 07 Part No. or Volume No. : 08 Contract No. : 10 Title and subtitle : Coulometry for the determination of uranium and plutonium: past and present 11 Collation : 34 p., 2 figs., 7 tabs. 13 Project No. : 20 Personal author(s) : M.K. Sharma; J.V. Kamat; A.S. Ambolikar; J.S. Pillai; S.K. Aggarwal 21 Affiliation of author(s) : Fuel Chemistry Division, Bhabha Atomic Research Centre, Mumbai 22 Corporate author(s) : Bhabha Atomic Research Centre, Mumbai - 400 085 23 Originating unit : Fuel Chemistry Division, BARC, Mumbai 24 Sponsor(s) Name : Department of Atomic Energy Type : Government Contd... BARC/2012/E/001 30 Date of submission : December 2011 31 Publication/Issue date : January 2012 40 Publisher/Distributor : Head, Scientific Information Resource Division, Bhabha Atomic Research Centre, Mumbai 42 Form of distribution : Hard copy 50 Language of text : English 51 Language of summary : English, Hindi 52 No. -

Plutonium Analysis from Controlled-Potential Coulometry for the Certification of the MP3 Standard Material
P5_14 Plutonium analysis from controlled-potential coulometry for the certification of the MP3 standard material. A. Ruas, V. Dalier, J. Pivato CEA-Marcoule BP 17171 30207 Bagnols-sur-Cèze Cedex, France [email protected] Abstract – For contributing to the certification of the new metal plutonium reference material (MP3), controlled-potential coulometry (CPC) has many advantages: it is a high accuracy absolute chemical analysis technique. Many studies are now conducted on plutonium solutions, to improve the operating conditions and the current apparatus, for mass determination with a precision of 0.1%. The different experimental preliminary results are discussed and the apparatus described. The coulometry cell assembly comprises a motor connected to a stirrer designed to prevent splashing, an inlet tube for inert gas, three electrodes, and a thermocouple for measuring the temperature. The measuring system includes a potentiostat, a CPU, a calibrated current generator, a temperature indicator and a voltmeter, all maintained at a constant temperature. Current integration is made by electronic components, introduced in the potentiostat and the CPU. + − + INTRODUCTION Pu 4 + e ↔ Pu 3 (1) Experimental electrolysis is performed using a Coulometry is an assay method in which the metal electrode with a large surface area quantity of the element analyzed is determined (working electrode) immersed in the test by measuring a quantity of electricity; under solution. The quantity of electricity used for the certain conditions it is capable of providing a conversion is related to the quantity matter in very accurate determination of the plutonium solution by Faraday’s law of electrolysis: mass concentration. The advantage of this M method is that it is absolute and uses only small m = ⋅Q (2) masses of material. -

School of Engineering and Science to My Beloved Parents Abstract
Hybrid Organic-Inorganic Polyoxometalates Functionalized by Diorganotin Groups by Firasat Hussain A thesis submitted in partial ful¯llment of the requirements for the degree of Doctor of Philosophy Approved, Thesis Committee: Prof. Ulrich Kortz (Mentor), IUB Prof. Ryan M Richards, IUB Dr. Michael H Dickman, IUB Prof. Michael T Pope Georgetown University, U.S.A. Prof. Emmanuel Cadot Universit¶ede Versailles, France Date of defense: 19 May 2006 School of Engineering and Science To my beloved parents Abstract Polyoxometalates (POMs) are a well-known class of inorganic metal-oxygen clusters with an unmatched structural variety combined with a multitude of properties. The search for novel POMs is predominantly driven by exciting catalytic, medicinal, material science and bioscience applications. However, the mechanism of action of most polyoxoanions is not selective towards a speci¯c target. In order to improve selectivity it appears highly desirable to attach organic functionalities covalently to the surface of polyoxoanions. The hydrolytic stability of the Sn-C bond enables the synthesis of a novel class of polyoxoanions via attachment of organometallic functionalities based on Sn(IV) to the surface of lacunary polyoxoanion precursors. III III By reacting (CH3)2SnCl2 with Na9(®-XW9O33) (X = As , Sb ) in aqueous acidic medium leads to the formation of 2-D solid-state structures with inorganic and organic surface, which are rare examples of discrete polyoxoanions. (CsNa4f(Sn(CH3)2)3O(H2O)4 (¯-AsW9O33)g¢5H2O)1 (CsNa-1) and the isostructural (CsNa4[(Sn(CH3)2)3O(H2O)4( ¯-SbW9O33)]¢5H2O)1 (CsNa-2)It has been synthesized and characterized by multinu- clear NMR spectroscopy, FTIR spectroscopy and elemental analysis. -

Chronoamperometry
Chronoamperometry • Stationary electrode • Unstirred = mass transport by diffusion • Constant potential • Measure current vs time Theory assume Ox + n e- Red - both Ox and Red are soluble - reversible reaction (electrochemically) - potential set so reduction goes to completion at the electrode surface Components of output signal in Chronoamperometry IFar decreases because Ox used up at electrode surface and Ox is only replenished by diffusion Faradaic current (IFar) follows Cottrell equation I Capacitive current (Icap) (current) decays exponentially for a constant applied potential t (time) Icap is high as electrode capacitive layer charges up, then drops off Processes perturbing system can cause data to differ from Cottrell Equation 1) Capacitive Current – charging current is exponential as shown -kt Icap = e Note: Capacitive current decreases more rapidly than Faradaic current so at longer times the ratio IFar/Icap is larger 2) Occurrence of coupled chemical reactions e.g. Ox + n e- Red 2 Red A A + n e- B Affects the shape of the current-time curve Chronoamperometry Applications • Can measure concentration by measuring I vs conc. at any fixed time • Can analyze the shape of the current-time curve in order to study coupled chemical reactions • There are better ways to do both of these with more modern techniques • Chronoamperometry is important because it is a fundamental method on which other techniques are based Chronopotentiometry • Stationary electrode • Unstirred = mass transport by diffusion • Constant current applied between -
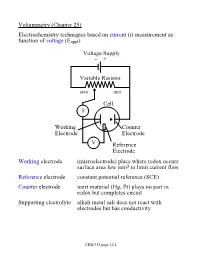
Voltammetry (Chapter 25) Electrochemistry Techniques Based on Current (I) Measurement As Function of Voltage (Eappl)
Voltammetry (Chapter 25) Electrochemistry techniques based on current (i) measurement as function of voltage (Eappl) Voltage Supply - + Variable Resistor max min Cell I Working Counter Electrode Electrode V Reference Electrode Working electrode (microelectrode) place where redox occurs surface area few mm2 to limit current flow Reference electrode constant potential reference (SCE) Counter electrode inert material (Hg, Pt) plays no part in redox but completes circuit Supporting electrolyte alkali metal salt does not react with electrodes but has conductivity CEM 333 page 12.1 Why not use 2 electrodes? OK in potentiometry - very small currents. Now, want to measure current (larger=better) but • potential drops when current is taken from electrode (IR drop) • must minimize current withdrawn from reference electrode surface Potentiostat (voltage source) drives cell • supplies whatever voltage needed between working and counter electrodes to maintain specific voltage between working and reference electrode NOTE: • Almost all current carried between working and counter electrodes • Voltage measured between working and reference electrodes • Analyte dissolved in cell not at electrode surface! CEM 333 page 12.2 Excitation signals (Fig 25-2) CEM 333 page 12.3 Microelectrodes C, Au, Pt, Hg each useful in certain solutions/voltage ranges Fig 25-4 At -ve limit, oxidation of water + - 2H2O ® 4H + O2(g) + 4e At +ve limit, reduction of water - - 2H2O + 2e ® H2 + 2OH CEM 333 page 12.4 Varies with material/solution due to different overpotentials Overpotential