Digital Expression Profiling of the Compartmentalized Translatome of Purkinje Neurons
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
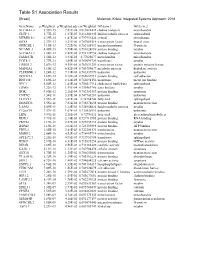
Table S1 Association Results
GeneName p.Weighted p.Weighted.adj cor.Weighted GO.term.1 GO.term.2 SLC44A1.2 3.55E-15 7.81E-08 0.819822422 choline transport mitochondria GLTP.1 5.77E-15 1.27E-07 0.816302445 lipid metabolic process sphingolipid MTMR10.1 6.39E-14 1.41E-06 0.797951424 cytosol phosphatase SOX8 2.37E-13 5.21E-06 0.787085514 transcription factor neural crest GPRC5B.1 4.19E-13 9.22E-06 0.782154937 integral membrane G-protein NCAM1.3 4.45E-13 9.79E-06 0.781624896 protein binding myelin SLC44A1.1 1.28E-12 2.82E-05 0.772132954 choline transport mitochondria FAM107B 1.56E-12 3.43E-05 0.77025677 mitochondria mitochondria UGT8.1 1.77E-12 3.89E-05 0.769099729 transferase myelin ERBB3.2 2.07E-12 4.55E-05 0.767631259 transcription factor protein tyrosine kinase MAN2A1 3.10E-12 6.82E-05 0.763759677 metabolic process hydrolase activity PLEKHH1.1 3.24E-12 7.13E-05 0.763337879 unknown unknown DOCK5.3 3.69E-12 8.12E-05 0.762087913 protein binding cell adhesion RNF130 3.69E-12 8.12E-05 0.762094156 membrane metal ion binding NPC1 6.50E-12 1.43E-04 0.756517114 cholesterol trafficking sphingolipid ERMN 7.22E-12 1.59E-04 0.755469786 actin binding myelin BOK 9.80E-12 2.16E-04 0.752383357 protein binding apoptosis CNTN2 1.54E-11 3.39E-04 0.747743281 unknown unknown ELOVL1 1.55E-11 3.41E-04 0.74764744 fatty acid sphingolipid DBNDD2 3.55E-11 7.81E-04 0.738878658 protein binding neuron projection LASS2 5.09E-11 1.12E-03 0.734954024 lipid metabolic process myelin C12orf34 7.57E-11 1.67E-03 0.730528911 unknown unknown LIPA 9.59E-11 2.11E-03 0.72786111 fatty acid glycerolipid metabolic -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/079361 Al 25 April 2019 (25.04.2019) W 1P O PCT (51) International Patent Classification: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, C12Q 1/68 (2018.01) A61P 31/18 (2006.01) DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, C12Q 1/70 (2006.01) HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, (21) International Application Number: MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, PCT/US2018/056167 OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, (22) International Filing Date: SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 16 October 2018 (16. 10.2018) TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every kind of regional protection available): ARIPO (BW, GH, (26) Publication Language: English GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, (30) Priority Data: UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, 62/573,025 16 October 2017 (16. 10.2017) US TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, DK, EE, ES, FI, FR, GB, GR, HR, HU, ΓΕ , IS, IT, LT, LU, LV, (71) Applicant: MASSACHUSETTS INSTITUTE OF MC, MK, MT, NL, NO, PL, PT, RO, RS, SE, SI, SK, SM, TECHNOLOGY [US/US]; 77 Massachusetts Avenue, TR), OAPI (BF, BJ, CF, CG, CI, CM, GA, GN, GQ, GW, Cambridge, Massachusetts 02139 (US). -

Transposable Elements Resistant to Epigenetic Resetting in the Human Germline Are Epigenetic Hotspots for Development and Disease
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.19.998930; this version posted March 20, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Transposable elements resistant to epigenetic resetting in the human germline are epigenetic hotspots for development and disease Sabine Dietmann1,2,3,11*, Michael J Keogh4,5,11, Walfred Tang2, Erna Magnusdottir6, Toshihiro Kobayashi7,8, Patrick F Chinnery5,9 and M. Azim Surani 1,2,10* ** 1. Wellcome Trust – Medical Research Council Stem Cell Institute, University of Cambridge, UK 2. Wellcome Trust/Cancer Research UK Gurdon Institute, University of Cambridge, Cambridge, UK 3. Department of Developmental Biology and Division of Nephrology, Washington University School of Medicine, St. Louis, MO, USA 4. Institute of Genetic Medicine, Newcastle University, Newcastle Upon Tyne, UK 5. MRC-Mitochondrial Biology Unit, University of Cambridge, Cambridge, UK 6. Department of Biomedical Science and Anatomy, Faculty of Medicine, University of Iceland, Reykjavik, Iceland 7. Section of Mammalian Transgenesis, Center for Genetic Analysis of Behavior, National Institute for Physiological Sciences, Okazaki, Japan 8. Department of Physiological Sciences, The Graduate University of Advanced Studies, Okazaki, Japan 9. Department of Clinical Neurosciences, Cambridge Biomedical Campus, University of Cambridge, Cambridge, UK 10. Department of Physiology and Development of Neuroscience, University of Cambridge, UK 11. These authors contributed equally * Co-corresponding authors; [email protected]; [email protected] ** lead contact:[email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.03.19.998930; this version posted March 20, 2020. -

Early Growth Response 1 Regulates Hematopoietic Support and Proliferation in Human Primary Bone Marrow Stromal Cells
Hematopoiesis SUPPLEMENTARY APPENDIX Early growth response 1 regulates hematopoietic support and proliferation in human primary bone marrow stromal cells Hongzhe Li, 1,2 Hooi-Ching Lim, 1,2 Dimitra Zacharaki, 1,2 Xiaojie Xian, 2,3 Keane J.G. Kenswil, 4 Sandro Bräunig, 1,2 Marc H.G.P. Raaijmakers, 4 Niels-Bjarne Woods, 2,3 Jenny Hansson, 1,2 and Stefan Scheding 1,2,5 1Division of Molecular Hematology, Department of Laboratory Medicine, Lund University, Lund, Sweden; 2Lund Stem Cell Center, Depart - ment of Laboratory Medicine, Lund University, Lund, Sweden; 3Division of Molecular Medicine and Gene Therapy, Department of Labora - tory Medicine, Lund University, Lund, Sweden; 4Department of Hematology, Erasmus MC Cancer Institute, Rotterdam, the Netherlands and 5Department of Hematology, Skåne University Hospital Lund, Skåne, Sweden ©2020 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2019.216648 Received: January 14, 2019. Accepted: July 19, 2019. Pre-published: August 1, 2019. Correspondence: STEFAN SCHEDING - [email protected] Li et al.: Supplemental data 1. Supplemental Materials and Methods BM-MNC isolation Bone marrow mononuclear cells (BM-MNC) from BM aspiration samples were isolated by density gradient centrifugation (LSM 1077 Lymphocyte, PAA, Pasching, Austria) either with or without prior incubation with RosetteSep Human Mesenchymal Stem Cell Enrichment Cocktail (STEMCELL Technologies, Vancouver, Canada) for lineage depletion (CD3, CD14, CD19, CD38, CD66b, glycophorin A). BM-MNCs from fetal long bones and adult hip bones were isolated as reported previously 1 by gently crushing bones (femora, tibiae, fibulae, humeri, radii and ulna) in PBS+0.5% FCS subsequent passing of the cell suspension through a 40-µm filter. -

High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype
Supplementary Information High Constitutive Cytokine Release by Primary Human Acute Myeloid Leukemia Cells Is Associated with a Specific Intercellular Communication Phenotype Håkon Reikvam 1,2,*, Elise Aasebø 1, Annette K. Brenner 2, Sushma Bartaula-Brevik 1, Ida Sofie Grønningsæter 2, Rakel Brendsdal Forthun 2, Randi Hovland 3,4 and Øystein Bruserud 1,2 1 Department of Clinical Science, University of Bergen, 5020, Bergen, Norway 2 Department of Medicine, Haukeland University Hospital, 5021, Bergen, Norway 3 Department of Medical Genetics, Haukeland University Hospital, 5021, Bergen, Norway 4 Institute of Biomedicine, University of Bergen, 5020, Bergen, Norway * Correspondence: [email protected]; Tel.: +55-97-50-00 J. Clin. Med. 2019, 8, x 2 of 36 Figure S1. Mutational studies in a cohort of 71 AML patients. The figure shows the number of patients with the various mutations (upper), the number of mutations in for each patient (middle) and the number of main classes with mutation(s) in each patient (lower). 2 J. Clin. Med. 2019, 8, x; doi: www.mdpi.com/journal/jcm J. Clin. Med. 2019, 8, x 3 of 36 Figure S2. The immunophenotype of primary human AML cells derived from 62 unselected patients. The expression of the eight differentiation markers CD13, CD14, CD15, CD33, CD34, CD45, CD117 and HLA-DR was investigated for 62 of the 71 patients included in our present study. We performed an unsupervised hierarchical cluster analysis and identified four patient main clusters/patient subsets. The mutational profile for each f the 62 patients is also given (middle), no individual mutation of main class of mutations showed any significant association with any of the for differentiation marker clusters (middle). -

Direct Interaction Between Hnrnp-M and CDC5L/PLRG1 Proteins Affects Alternative Splice Site Choice
Direct interaction between hnRNP-M and CDC5L/PLRG1 proteins affects alternative splice site choice David Llères, Marco Denegri, Marco Biggiogera, Paul Ajuh, Angus Lamond To cite this version: David Llères, Marco Denegri, Marco Biggiogera, Paul Ajuh, Angus Lamond. Direct interaction be- tween hnRNP-M and CDC5L/PLRG1 proteins affects alternative splice site choice. EMBO Reports, EMBO Press, 2010, 11 (6), pp.445 - 451. 10.1038/embor.2010.64. hal-03027049 HAL Id: hal-03027049 https://hal.archives-ouvertes.fr/hal-03027049 Submitted on 26 Nov 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. scientificscientificreport report Direct interaction between hnRNP-M and CDC5L/ PLRG1 proteins affects alternative splice site choice David Lle`res1*, Marco Denegri1*w,MarcoBiggiogera2,PaulAjuh1z & Angus I. Lamond1+ 1Wellcome Trust Centre for Gene Regulation & Expression, College of Life Sciences, University of Dundee, Dundee, UK, and 2LaboratoriodiBiologiaCellulareandCentrodiStudioperl’IstochimicadelCNR,DipartimentodiBiologiaAnimale, Universita’ di Pavia, Pavia, Italy Heterogeneous nuclear ribonucleoprotein-M (hnRNP-M) is an and affect the fate of heterogeneous nuclear RNAs by influencing their abundant nuclear protein that binds to pre-mRNA and is a structure and/or by facilitating or hindering the interaction of their component of the spliceosome complex. -
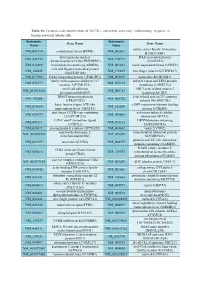
Systematic Name Gene Name Systematic Name Gene Name NM 001710 Complement Factor B(CFB) NM 052831 Solute Carrier Family 18 Member
Table S1: Genome-wide identification of SGLT2i`s interaction with early inflammatory response in human proximal tubular cells. Systematic Systematic Gene Name Gene Name Name Name solute carrier family 18 member NM_001710 complement factor B(CFB) NM_052831 B1(SLC18B1) heterogeneous nuclear DAZ associated protein NM_031372 NM_170711 ribonucleoprotein D like(HNRNPDL) 1(DAZAP1) NM_014299 bromodomain containing 4(BRD4) NM_001261 cyclin dependent kinase 9(CDK9) cilia and flagella associated protein NM_182628 NM_178835 zinc finger protein 827(ZNF827) 100(CFAP100) NM_017906 PAK1 interacting protein 1(PAK1IP1) NM_024015 homeobox B4(HOXB4) family with sequence similarity 167 ankyrin repeat and LEM domain NM_053279 NM_015114 member A(FAM167A) containing 2(ANKLE2) small cell adhesion ARP3 actin related protein 3 NM_001031628 NM_005721 glycoprotein(SMAGP) homolog(ACTR3) TRAF3 interacting protein actin related protein 2/3 complex NM_147686 NM_005720 2(TRAF3IP2) subunit 1B(ARPC1B) basic leucine zipper ATF-like cAMP responsive element binding NM_018664 NM_182898 transcription factor 3(BATF3) protein 5(CREB5) zinc finger CCCH-type containing activation induced cytidine NM_025079 NM_020661 12A(ZC3H12A) deaminase(AICDA) C-X-C motif chemokine ligand DENN domain containing NM_001511 NM_015213 1(CXCL1) 5A(DENND5A) NM_025072 prostaglandin E synthase 2(PTGES2) NM_004665 vanin 2(VNN2) superoxide dismutase 2, mitochondrial ribosomal protein NM_001024465 NM_016070 mitochondrial(SOD2) S23(MRPS23) jumonji and AT-rich interaction NM_033199 urocortin 2(UCN2) NM_004973 -

Pcagopromoter - an R Package for Biological and Regulatory Interpretation of Principal Components in Genome-Wide Gene Expression Data
Roskilde University pcaGoPromoter - An R Package for Biological and Regulatory Interpretation of Principal Components in Genome-Wide Gene Expression Data Hansen, Morten; Gerds, Thomas Alexander; Sedelin, Jacob Benedikt; Troelsen, Jesper; Olsen, Jørgen Published in: P L o S One DOI: 10.1371/journal.pone.0032394 Publication date: 2012 Document Version Publisher's PDF, also known as Version of record Citation for published version (APA): Hansen, M., Gerds, T. A., Sedelin, J. B., Troelsen, J., & Olsen, J. (2012). pcaGoPromoter - An R Package for Biological and Regulatory Interpretation of Principal Components in Genome-Wide Gene Expression Data. P L o S One, 7(2), e32394. https://doi.org/10.1371/journal.pone.0032394 General rights Copyright and moral rights for the publications made accessible in the public portal are retained by the authors and/or other copyright owners and it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights. • Users may download and print one copy of any publication from the public portal for the purpose of private study or research. • You may not further distribute the material or use it for any profit-making activity or commercial gain. • You may freely distribute the URL identifying the publication in the public portal. Take down policy If you believe that this document breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 28. Sep. 2021 -

In Cancer Cells by Heat Shock Stimulation
583-593.qxd 19/7/2010 11:13 Ì ™ÂÏ›‰·583 INTERNATIONAL JOURNAL OF ONCOLOGY 37: 583-593, 2010 583 Induction of HITS, a newly identified family with sequence similarity 107 protein (FAM107B), in cancer cells by heat shock stimulation HIDEO NAKAJIMA1, YASUHITO ISHIGAKI2, QI-SHENG XIA1, TAKAYUKI IKEDA3, YOSHINO YOSHITAKE3, HIDETO YONEKURA3, TAKAYUKI NOJIMA4, TAKUJI TANAKA4, HISANORI UMEHARA5, NAOHISA TOMOSUGI2, TAKANOBU TAKATA2, TAKEO SHIMASAKI1, NAOKI NAKAYA1, ITARU SATO1, KAZUYUKI KAWAKAMI6, KEITA KOIZUMI7, TOSHINARI MINAMOTO6 and YOSHIHARU MOTOO1 1Department of Medical Oncology, 2Medical Research Institute and Departments of 3Biochemistry, 4Pathology and 5Hematology and Immunology, Kanazawa Medical University, 1-1 Daigaku, Uchinada, Kahoku, Ishikawa 920-0293; 6Division of Translational and Clinical Oncology, Cancer Research Institute and 7Department of Biophysical Genetics, Graduate School of Medical Science, Kanazawa University, 13-1 Takara-machi, Kanazawa, Ishikawa 920-0934, Japan Received March 31, 2010; Accepted May 14, 2010 DOI: 10.3892/ijo_00000707 Abstract. The Family with sequence similarity 107 (FAM107) Introduction possesses an N-terminal domain of unknown function (DUF1151) that is highly conserved beyond species. In human, The Family with sequence similarity 107 (FAM107) possesses FAM107A termed TU3A/DRR1 has been reported as a can- an N-terminal domain of unknown function (DUF1151) that didate tumor suppressor gene which expression is down- is conserved beyond species including mammalian, Xenopus, regulated in several types of cancer, however no studies have fish and Drosophila, and shows no homology match to investigated the other family protein, FAM107B. In the other functional conserved domains (http://www.ncbi.nlm. present study, we designated FAM107B as heat shock- nih.gov/structure/cdd/cdd.shtml). -

LADON, a Natural Antisense Transcript of NODAL, Promotes Metastasis in Melanoma by Repressing NDRG1
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.09.032375; this version posted April 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. LADON, a natural antisense transcript of NODAL, promotes metastasis in melanoma by repressing NDRG1 Dutriaux Annie1, Diazzi Serena1, Caburet Sandrine2, Bresesti Chiara1, Hardouin Sylvie1, Deshayes Frédérique3, Collignon Jérôme1,* and Flagiello Domenico1* 1: Université de Paris, CNRS, Institut Jacques Monod, Regulation of Cell-Fate Specification team, Paris 75013, France 2: Université de Paris, CNRS, Institut Jacques Monod, Molecular Oncology and Ovarian Pathologies team, Paris 75013, France 3: Université de Paris, CNRS, Institut Jacques Monod, Morphogenesis, Homeostasis and Pathologies team, Paris 75013, France *Correspondence: [email protected]; [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.04.09.032375; this version posted April 11, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Summary The TGFb family member NODAL, primarily known for its role during embryonic development, has also been associated with tumor progression in a number of cancers. Some of the evidence supporting its involvement in melanoma has appeared contradictory, suggesting that NODAL in this context might rely on a non-canonical mode of signaling. To investigate this possibility we studied how a deletion of NODAL affected cell behavior in a metastatic melanoma cell line. The mutation does prevent melanoma cells from acquiring an invasive behavior. -

Transcriptome Profiling of Cleft Palate Intgf-Beta3 Knockout Mice Alleles: RNA-SEQ Analysis of TGF-Beta3 Mice" (2017)
University of Nebraska Medical Center DigitalCommons@UNMC Theses & Dissertations Graduate Studies Fall 12-15-2017 Transcriptome Profiling of Cleft alateP inTGF-beta3 Knockout Mice Alleles: RNA-SEQ Analysis of TGF-beta3 Mice Kelsey White University of Nebraska Medical Center Follow this and additional works at: https://digitalcommons.unmc.edu/etd Part of the Bioinformatics Commons, and the Developmental Biology Commons Recommended Citation White, Kelsey, "Transcriptome Profiling of Cleft Palate inTGF-beta3 Knockout Mice Alleles: RNA-SEQ Analysis of TGF-beta3 Mice" (2017). Theses & Dissertations. 239. https://digitalcommons.unmc.edu/etd/239 This Thesis is brought to you for free and open access by the Graduate Studies at DigitalCommons@UNMC. It has been accepted for inclusion in Theses & Dissertations by an authorized administrator of DigitalCommons@UNMC. For more information, please contact [email protected]. TRANSCRIPTOME PROFILING OF CLEFT PALATE IN TGF-3 KNOCKOUT MICE ALLELES: RNA-SEQ ANALYSIS OF TGF-3 MICE By Kelsey Marie White, D.D.S. A THESIS Presented to the Faculty of the University of Nebraska Graduate College in Partial Fulfillment of Requirements for the Degree of Master of Science Medical Sciences Interdepartmental Area Graduate Program (Oral Biology) Under the Supervision of Ali Nawshad, Ph.D. University of Nebraska Medical Center Omaha, Nebraska December, 2017 Advisory Committee: Peter Giannini, D.D.S., M.S Ali Nawshad, Ph.D. S. Prem Premaraj, B.D.S., M.S., Ph. D. FRCD(C) Hasan Otu, Ph.D. i ACKNOWLEDGEMENTS First, I would like to sincerely thank my leader and mentor, Dr. Ali Nawshad, for the opportunity to work with and learn from him.