Mutations in COX7B Cause Microphthalmia with Linear Skin Lesions, an Unconventional Mitochondrial Disease
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genomic analysis of bone marrow failure and myelodysplastic syndromes reveals phenotypic and diagnostic complexity Michael Y. Zhang,1 Siobán B. Keel,2 Tom Walsh,3 Ming K. Lee,3 Suleyman Gulsuner,3 Amanda C. Watts,3 Colin C. Pritchard,4 Stephen J. Salipante,4 Michael R. Jeng,5 Inga Hofmann,6 David A. Williams,6,7 Mark D. Fleming,8 Janis L. Abkowitz,2 Mary-Claire King,3 and Akiko Shimamura1,9,10 M.Y.Z. and S.B.K. contributed equally to this work. 1Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, WA; 2Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 3Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; 4Department of Laboratory Medicine, University of Washington, Seattle, WA; 5Department of Pediatrics, Stanford Uni- versity School of Medicine, Stanford, CA; 6Division of Hematology/Oncology, Boston Children’s Hospital, Dana Farber Cancer Insti- tute, and Harvard Medical School, Boston, MA; 7Harvard Stem Cell Institute, Boston, MA; 8Department of Pathology, Boston Children’s Hospital, MA; 9Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 10Department of Pedi- atrics, University of Washington, Seattle, WA, USA ©2014 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2014.113456 Manuscript received on July 22, 2014. Manuscript accepted on September 15, 2014. Correspondence: [email protected] Supplementary Methods Genomics. Libraries were prepared in 96-well format with a Bravo liquid handling robot (Agilent Technologies). One to two micrograms of genomic DNA were sheared to a peak size of 150 bp using a Covaris E series instrument. -
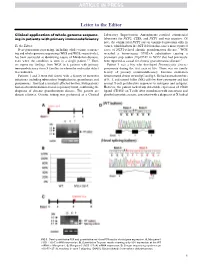
Clinical Application of Whole-Genome Sequencing in Patients with Primary
Letter to the Editor Clinical application of whole-genome sequenc- Laboratory Improvement Amendments–certified commercial ing in patients with primary immunodeficiency laboratory for NCF2, CYBA, and NCF1 and was negative. Of note, the commercial NCF1 screen examined mutations only in To the Editor: exon 2, which harbors the 2GT deletion that causes most reported Next-generation sequencing, including whole-exome sequenc- cases of NCF1-related chronic granulomatous disease.4 WGS ing and whole-genome sequencing (WES and WGS, respectively), revealed a homozygous 579G>A substitution causing a has been successful at identifying causes of Mendelian diseases, premature stop codon (Trp193X) in NCF1 that had previously even when the condition is seen in a single patient.1-3 Here, been reported as causal for chronic granulomatous disease.5 we report our findings from WGS in 6 patients with primary Patient 3 was a boy who developed Pneumocystis jiroveci immunodeficiency from 5 families in whom the molecular defect pneumonia during the first year of life. There was no family was unknown. history of primary immunodeficiency. Immune evaluation Patients 1 and 2 were full sisters with a history of recurrent demonstrated absent serum IgG and IgA. He had normal numbers infections, including tuberculous lymphadenitis, granulomas, and of B, T, and natural killer (NK) cells by flow cytometry and had pneumonias. They had a similarly affected brother. Both patients normal T-cell proliferative responses to mitogens and antigens. had an absent rhodamine-based respiratory burst, confirming the However, the patient lacked any detectable expression of CD40 diagnosis of chronic granulomatous disease. The parents are ligand (CD154) on T cells after stimulation with ionomycin and distant relatives. -

In Silico Tools for Splicing Defect Prediction: a Survey from the Viewpoint of End Users
© American College of Medical Genetics and Genomics REVIEW In silico tools for splicing defect prediction: a survey from the viewpoint of end users Xueqiu Jian, MPH1, Eric Boerwinkle, PhD1,2 and Xiaoming Liu, PhD1 RNA splicing is the process during which introns are excised and informaticians in relevant areas who are working on huge data sets exons are spliced. The precise recognition of splicing signals is critical may also benefit from this review. Specifically, we focus on those tools to this process, and mutations affecting splicing comprise a consider- whose primary goal is to predict the impact of mutations within the able proportion of genetic disease etiology. Analysis of RNA samples 5′ and 3′ splicing consensus regions: the algorithms used by different from the patient is the most straightforward and reliable method to tools as well as their major advantages and disadvantages are briefly detect splicing defects. However, currently, the technical limitation introduced; the formats of their input and output are summarized; prohibits its use in routine clinical practice. In silico tools that predict and the interpretation, evaluation, and prospection are also discussed. potential consequences of splicing mutations may be useful in daily Genet Med advance online publication 21 November 2013 diagnostic activities. In this review, we provide medical geneticists with some basic insights into some of the most popular in silico tools Key Words: bioinformatics; end user; in silico prediction tool; for splicing defect prediction, from the viewpoint of end users. Bio- medical genetics; splicing consensus region; splicing mutation INTRODUCTION TO PRE-mRNA SPLICING AND small nuclear ribonucleoproteins and more than 150 proteins, MUTATIONS AFFECTING SPLICING serine/arginine-rich (SR) proteins, heterogeneous nuclear ribo- Sixty years ago, the milestone discovery of the double-helix nucleoproteins, and the regulatory complex (Figure 1). -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients
Bone Marrow Failure SUPPLEMENTARY APPENDIX Genetic features of myelodysplastic syndrome and aplastic anemia in pediatric and young adult patients Siobán B. Keel, 1* Angela Scott, 2,3,4 * Marilyn Sanchez-Bonilla, 5 Phoenix A. Ho, 2,3,4 Suleyman Gulsuner, 6 Colin C. Pritchard, 7 Janis L. Abkowitz, 1 Mary-Claire King, 6 Tom Walsh, 6** and Akiko Shimamura 5** 1Department of Medicine, Division of Hematology, University of Washington, Seattle, WA; 2Clinical Research Division, Fred Hutchinson Can - cer Research Center, Seattle, WA; 3Department of Pediatric Hematology/Oncology, Seattle Children’s Hospital, WA; 4Department of Pedi - atrics, University of Washington, Seattle, WA; 5Boston Children’s Hospital, Dana Farber Cancer Institute, and Harvard Medical School, MA; 6Department of Medicine and Department of Genome Sciences, University of Washington, Seattle, WA; and 7Department of Laboratory Medicine, University of Washington, Seattle, WA, USA *SBK and ASc contributed equally to this work **TW and ASh are co-senior authors ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol. 2016.149476 Received: May 16, 2016. Accepted: July 13, 2016. Pre-published: July 14, 2016. Correspondence: [email protected] or [email protected] Supplementary materials Supplementary methods Retrospective chart review Patient data were collected from medical records by two investigators blinded to the results of genetic testing. The following information was collected: date of birth, transplant, death, and last follow-up, -

Mitoxplorer, a Visual Data Mining Platform To
mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, Cecilia Garcia-Perez, José Villaveces, Salma Gamal, Giovanni Cardone, Fabiana Perocchi, et al. To cite this version: Annie Yim, Prasanna Koti, Adrien Bonnard, Fabio Marchiano, Milena Dürrbaum, et al.. mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dy- namics and mutations. Nucleic Acids Research, Oxford University Press, 2020, 10.1093/nar/gkz1128. hal-02394433 HAL Id: hal-02394433 https://hal-amu.archives-ouvertes.fr/hal-02394433 Submitted on 4 Dec 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License Nucleic Acids Research, 2019 1 doi: 10.1093/nar/gkz1128 Downloaded from https://academic.oup.com/nar/advance-article-abstract/doi/10.1093/nar/gkz1128/5651332 by Bibliothèque de l'université la Méditerranée user on 04 December 2019 mitoXplorer, a visual data mining platform to systematically analyze and visualize mitochondrial expression dynamics and mutations Annie Yim1,†, Prasanna Koti1,†, Adrien Bonnard2, Fabio Marchiano3, Milena Durrbaum¨ 1, Cecilia Garcia-Perez4, Jose Villaveces1, Salma Gamal1, Giovanni Cardone1, Fabiana Perocchi4, Zuzana Storchova1,5 and Bianca H. -

Spectrum of Splicing Errors Caused by CHRNE Mutations Affecting Introns and Intron/Exon Boundaries K Ohno, a Tsujino, X-M Shen, M Milone, a G Engel
1of5 ONLINE MUTATION REPORT J Med Genet: first published as 10.1136/jmg.2004.026682 on 1 August 2005. Downloaded from Spectrum of splicing errors caused by CHRNE mutations affecting introns and intron/exon boundaries K Ohno, A Tsujino, X-M Shen, M Milone, A G Engel ............................................................................................................................... J Med Genet 2005;42:e53 (http://www.jmedgenet.com/cgi/content/full/42/8/e53). doi: 10.1136/jmg.2004.026682 Patients Background: Mutations in CHRNE, the gene encoding the Patients 1–5 (respectively a 59 year old woman, a 23 year old muscle nicotinic acetylcholine receptor e subunit, cause man, a 2.5 year old girl, a 6 year old boy, and a 44 year old congenital myasthenic syndromes. Only three of the eight man) have moderate to severe myasthenic symptoms that intronic splice site mutations of CHRNE reported to date have have been present since birth or infancy, decremental EMG had their splicing consequences characterised. responses, and no AChR antibodies. All respond partially to Methods: We analysed four previously reported and five pyridostigmine. Patient 4 underwent an intercostal muscle novel splicing mutations in CHRNE by introducing the entire biopsy for diagnosis, which showed severe endplate AChR normal and mutant genomic CHRNEs into COS cells. deficiency (6% of normal) and compensatory expression of Results and conclusions: We found that short introns (82– the fetal c-AChR at the endplate. 109 nucleotides) favour intron retention, whereas medium to long introns (306–1210 nucleotides) flanking either or both Construction of CHRNE clones for splicing analysis sides of an exon favour exon skipping. Two mutations are of To examine the consequences of the identified splice site particular interest. -

Background Splicing and Genetic Disease
Background splicing and genetic disease Diana Alexieva Imperial College London Yi Long Imperial College London Rupa Sarkar Imperial College London Hansraj Dhayan Imperial College London Emmanuel Bruet Imperial College London Robert Winston Imperial College London Igor Vorechovsky University of Southampton Leandro Castellano University of Sussex Nick Dibb ( [email protected] ) Imperial College London Research Article Keywords: background splicing, splice site mutations, cryptic splice sites, exon skipping, pseudoexons, recursive splicing, spliceosomal mutations, splicing therapy, BRCA1, BRCA1, DMD Posted Date: October 15th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-92665/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/23 Abstract We report that low level background splicing by normal genes can be used to predict the likely effect of splicing mutations upon cryptic splice site activation and exon skipping, with emphasis on the DBASS databases, BRCA1, BRCA2 and DMD. In addition we show that background RNA splice sites are also involved in pseudoexon formation, recursive splicing and aberrant splicing in cancer. We discuss how background splicing information might inform splicing therapy. Introduction We previously established that cryptic splices sites (css) are already active, albeit at very low levels, in normal genes. We did this by using EST data to identify rare splice sites and then compared their positions to known css that are activated in human disease (1). However, this approach was limited to a minority of genes for which there was sucient EST sequence data. Since that time a large amount of RNA-sequencing data has been deposited, which we reasoned would strongly increase the power of css prediction. -

Tissue-And Condition-Specific Isoforms of Mammalian Cytochrome
Hindawi Oxidative Medicine and Cellular Longevity Volume 2017, Article ID 1534056, 19 pages https://doi.org/10.1155/2017/1534056 Review Article Tissue- and Condition-Specific Isoforms of Mammalian Cytochrome c Oxidase Subunits: From Function to Human Disease 1 1 1 2 3 Christopher A. Sinkler, Hasini Kalpage, Joseph Shay, Icksoo Lee, Moh H. Malek, 1 1 Lawrence I. Grossman, and Maik Hüttemann 1Center for Molecular Medicine and Genetics, Wayne State University School of Medicine, Detroit, MI 48201, USA 2College of Medicine, Dankook University, Cheonan-si, Chungcheongnam-do 31116, Republic of Korea 3Department of Health Care Sciences, Eugene Applebaum College of Pharmacy and Health Sciences, Wayne State University, Detroit, MI 48201, USA Correspondence should be addressed to Maik Hüttemann; [email protected] Received 2 February 2017; Accepted 29 March 2017; Published 16 May 2017 Academic Editor: Ryuichi Morishita Copyright © 2017 Christopher A. Sinkler et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Cytochrome c oxidase (COX) is the terminal enzyme of the electron transport chain and catalyzes the transfer of electrons from cytochrome c to oxygen. COX consists of 14 subunits, three and eleven encoded, respectively, by the mitochondrial and nuclear DNA. Tissue- and condition-specific isoforms have only been reported for COX but not for the other oxidative phosphorylation complexes, suggesting a fundamental requirement to fine-tune and regulate the essentially irreversible reaction catalyzed by COX. This article briefly discusses the assembly of COX in mammals and then reviews the functions of the six nuclear-encoded COX subunits that are expressed as isoforms in specialized tissues including those of the liver, heart and skeletal muscle, lung, and testes: COX IV-1, COX IV-2, NDUFA4, NDUFA4L2, COX VIaL, COX VIaH, COX VIb-1, COX VIb-2, COX VIIaH, COX VIIaL, COX VIIaR, COX VIIIH/L, and COX VIII-3. -

Anti-COX7B Antibody
03/19 Anti-COX7B Antibody CATALOG NO.: A1756-100 100 μl. BACKGROUND DESCRIPTION: Cytochrome c oxidase (COX), the terminal component of the mitochondrial respiratory chain, catalyzes the electron transfer from reduced cytochrome c to oxygen. This component is a heteromeric complex consisting of 3 catalytic subunits encoded by mitochondrial genes and multiple structural subunits encoded by nuclear genes. The mitochondrial- encoded subunits function in electron transfer, and the nuclear-encoded subunits may function in the regulation and assembly of the complex. This nuclear gene encodes subunit VIIb, which is highly similar to bovine COX VIIb protein and is found in all tissues. This gene may have several pseudogenes on chromosomes 1, 2, 20 and 22. ALTERNATE NAMES: Cytochrome c oxidase polypeptide VIIb, Cytochrome c oxidase subunit 7B, Cytochrome c oxidase subunit 7B mitochondrial, APLCC. ANTIBODY TYPE: Polyclonal CONCENTRATION: 0.3 mg/ml HOST/ISOTYPE: Rabbit / IgG. IMMUNOGEN: Recombinant protein of human COX7B. MOLECULAR WEIGHT: 9 kDa. PURIFICATION: Affinity purification. FORM: Liquid. FORMULATION: PBS with 0.05% sodium azide, 50% glycerol, PH7.3 SPECIES REACTIVITY: Rat. Human. STORAGE CONDITIONS: Store at -20°C. Avoid freeze / thaw cycles. APPLICATIONS AND USAGE: WB 1:500-1:2000, IHC 1:50-1:200. : IHC staining of paraffin-embedded Human colon cancer using Anti-COX7B Antibody. 155 S. Milpitas Blvd., Milpitas, CA 95035 USA | T: (408)493-1800 F: (408)493-1801 | www.biovision.com | [email protected] 03/19 : Western Blot analysis of 293T cell using Anti-COX7B Antibody. RELATED PRODUCTS: Cox-3 Antibody (3687). Cyclooxygenase (COX) Activity Assay Kit (Fluorometric) (K549). TFPI Antibody (3379). -

COX7B Antibody
Product Datasheet COX7B Antibody Catalog No: #35581 Orders: [email protected] Description Support: [email protected] Product Name COX7B Antibody Host Species Rabbit Clonality Polyclonal Purification Antigen affinity purification. Applications WB IHC Species Reactivity Hu Specificity The antibody detects endogenous levels of total COX7B protein. Immunogen Type Recombinant Protein Immunogen Description Fusion protein corresponding to a region derived from internal residues of human Cytochrome c oxidase subunit VIIb Target Name COX7B Other Names APLCC Accession No. Swiss-Prot#: P24311NCBI Gene ID: 1349Gene Accssion: BC018386 SDS-PAGE MW 9kd Concentration 1.8mg/ml Formulation Rabbit IgG in pH7.4 PBS, 0.05% NaN3, 40% Glycerol. Storage Store at -20°C Application Details Western blotting: 1:500-1:2000 Immunohistochemistry: 1:50-1:200 Images Gel: 15%+10%SDS-PAGE Lysate: 50ug 293T cell Primary antibody: 1/700 dilution Secondary antibody dilution: 1/8000 Exposure time: 30 seconds Address: 8400 Baltimore Ave., Suite 302, College Park, MD 20740, USA http://www.sabbiotech.com 1 Immunohistochemical analysis of paraffin-embedded Human colon cancer tissue using #35581 at dilution 1/60. Background Cytochrome c oxidase (COX), the terminal component of the mitochondrial respiratory chain, catalyzes the electron transfer from reduced cytochrome c to oxygen. This component is a heteromeric complex consisting of 3 catalytic subunits encoded by mitochondrial genes and multiple structural subunits encoded by nuclear genes. The mitochondrially-encoded subunits function in electron transfer, and the nuclear-encoded subunits may function in the regulation and assembly of the complex. This nuclear gene encodes subunit VIIb, which is highly similar to bovine COX VIIb protein and is found in all tissues. -

Mouse Cox6c Conditional Knockout Project (CRISPR/Cas9)
https://www.alphaknockout.com Mouse Cox6c Conditional Knockout Project (CRISPR/Cas9) Objective: To create a Cox6c conditional knockout Mouse model (C57BL/6J) by CRISPR/Cas-mediated genome engineering. Strategy summary: The Cox6c gene (NCBI Reference Sequence: NM_053071 ; Ensembl: ENSMUSG00000014313 ) is located on Mouse chromosome 15. 4 exons are identified, with the ATG start codon in exon 2 and the TGA stop codon in exon 3 (Transcript: ENSMUST00000014457). Exon 2 will be selected as conditional knockout region (cKO region). Deletion of this region should result in the loss of function of the Mouse Cox6c gene. To engineer the targeting vector, homologous arms and cKO region will be generated by PCR using BAC clone RP23-97E5 as template. Cas9, gRNA and targeting vector will be co-injected into fertilized eggs for cKO Mouse production. The pups will be genotyped by PCR followed by sequencing analysis. Note: Exon 2 starts from about 100% of the coding region. The knockout of Exon 2 will result in frameshift of the gene. The size of intron 1 for 5'-loxP site insertion: 840 bp, and the size of intron 2 for 3'-loxP site insertion: 1741 bp. The size of effective cKO region: ~617 bp. The cKO region does not have any other known gene. Page 1 of 8 https://www.alphaknockout.com Overview of the Targeting Strategy Wildtype allele gRNA region 5' gRNA region 3' 1 2 3 4 Targeting vector Targeted allele Constitutive KO allele (After Cre recombination) Legends Homology arm Exon of mouse Cox6c cKO region loxP site Page 2 of 8 https://www.alphaknockout.com Overview of the Dot Plot Window size: 10 bp Forward Reverse Complement Sequence 12 Note: The sequence of homologous arms and cKO region is aligned with itself to determine if there are tandem repeats.