The Validity of Classical Nucleation Theory and Its Application to Dislocation Nucleation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Colloidal Crystallization: Nucleation and Growth
Colloidal Crystallization: Nucleation and Growth Dave Weitz Harvard Urs Gasser Konstanz Andy Schofield Edinburgh Rebecca Christianson Harvard Peter Pusey Edinburgh Peter Schall Harvard Frans Spaepen Harvard Eric Weeks Emory John Crocker UPenn •Colloids as model systems – soft materials •Nucleation and growth of colloidal crystals •Defect growth in crystals •Binary Alloy Colloidal Crystals NSF, NASA http://www.deas.harvard.edu/projects/weitzlab EMU 6/7//04 Colloidal Crystals Colloids 1 nm - 10 µm solid particles in a solvent Ubiquitous ink, paint, milk, butter, mayonnaise, toothpaste, blood Suspensions can act like both liquid and solid Modify flow properties Control: Size, uniformity, interactions Colloidal Particles •Solid particles in fluid •Hard Spheres •Volume exclusion •Stability: •Short range repulsion •Sometimes a slight charge Colloid Particles are: •Big Slow •~ a ~ 1 micron • τ ~ a2/D ~ ms to sec •Can “see” them •Follow individual particle dynamics Model: Colloid Æ Atom Phase Behavior Colloids Atomic Liquids U U s s Hard Spheres Leonard-Jones Osmotic Pressure Attraction Centro-symmetric Centro-symmetric Thermalized: kBT Thermalized: kBT Gas Gas Liquid Density Liquid Solid Solid Phase behavior is similar Hard Sphere Phase Diagram Volume Fraction Controls Phase Behavior liquid - crystal liquid coexistence crystal 49% 54% 63% 74% φ maximum packing maximum packing φRCP≈0.63 φHCP=0.74 0 Increase φ => Decrease Temperature F = U - TS Entropy Drives Crystallization 0 Entropy => Free Volume F = U - TS Disordered: Ordered: •Higher configurational -

INTERNATIONAL INSTITUTE of BENGAL and HIMALAYAN BASINS 10 Evans Hall, University of California at Berkeley Berkeley, California
INTERNATIONAL INSTITUTE OF BENGAL AND HIMALAYAN BASINS 10 Evans Hall, University of California at Berkeley BERKELEY, CALIFORNIA The International Institute of the Bengal and Himalayan Basins PEACE July 20, 2013 TOWNES AND TAGORE FOURTH ANNUAL SEMINAR ON THE GLOBAL WATER CRISIS 1:30 – 2:00 PM RECEPTION/MIXER 2:00 – 2:15 PM POETRY / SONG Mamade Kadreebux Sushmita Ghosh 2:15-4:00 PM SEMINAR INTRODUCTION Rosalie Say Welcome Founder’s Introduction: Mamade Kadreebux Welcome and Prefatory Remarks, Rash B. Ghosh, PhD, Founder, IIBHB SPECIAL WORDS FROM FRIENDS & WELL-WISHERS OF PROFESSOR CHARLES TOWNES 2:45 – 4:00 PM SESSION ONE The Convergence of Science and Spirituality David Presti, PhD, Professor, Molecular Cell Biology, UC Berkeley Water Budget Estimation and Water Management in the Mekong River Basin Jeanny Wang, President/Sr. Environmental Engineer, EcoWang Ltd. Sand from Newton’s Seashore: Introduction of Dr. Charles H. Townes John Paulin, PhD, Technical Writer and Editor, IIBHB Chief Guest Address: Vivekananda and a Vision for the South Asia, the US, and our Planet Charles H. Townes, PhD, 1964 Nobel Laureate in Physics, 1999 Rabindranath Tagore Award Recipient, and 2005 Templeton Prize Awardee Q & A 4:30 - 6:30 SESSION TWO INTRODUCTION OF KEYNOTE SPEAKER Derek Whitworth, PhD, President, IIBHB Keynote Address Steven Chu, 1997 Nobel Laureate in Physics and former U.S. Secretary of Energy Reducing the Impact of Toxics in Drinking Water Resources Rash B. Ghosh, PhD, Founder, IIBHB Special Presentation: How Advances in Science are Made. Douglas Osheroff, PhD, 1996 Nobel Laureate in Physics Q & A SUMMARY AND CONCLUDING REMARKS Sterling Bunnel, MD, IIBHB Former President and Advisor ACKNOWLEDGEMENT Master of Ceremonies Rosalie Say Professor Charles Hard Townes was born in 1915 and invented the microwave laser, or maser, in 1953 while at Columbia University. -

See the Scientific Petition
May 20, 2016 Implement the Endangered Species Act Using the Best Available Science To: Secretary Sally Jewell and Secretary Penny Prtizker We, the under-signed scientists, recommend the U.S. government place species conservation policy on firmer scientific footing by following the procedure described below for using the best available science. A recent survey finds that substantial numbers of scientists at the U.S. Fish and Wildlife Service (FWS) and the National Oceanic and Atmospheric Administration believe that political influence at their agency is too high.i Further, recent species listing and delisting decisions appear misaligned with scientific understanding.ii,iii,iv,v,vi For example, in its nationwide delisting decision for gray wolves in 2013, the FWS internal review failed the best science test when reviewed by an independent peer-review panel.vii Just last year, a FWS decision not to list the wolverine ran counter to the opinions of agency and external scientists.viii We ask that the Departments of the Interior and Commerce make determinations under the Endangered Species Actix only after they make public the independent recommendations from the scientific community, based on the best available science. The best available science comes from independent scientists with relevant expertise who are able to evaluate and synthesize the available science, and adhere to standards of peer-review and full conflict-of-interest disclosure. We ask that agency scientific recommendations be developed with external review by independent scientific experts. There are several mechanisms by which this can happen; however, of greatest importance is that an independent, external, and transparent science-based process is applied consistently to both listing and delisting decisions. -

Entropy and the Tolman Parameter in Nucleation Theory
entropy Article Entropy and the Tolman Parameter in Nucleation Theory Jürn W. P. Schmelzer 1 , Alexander S. Abyzov 2 and Vladimir G. Baidakov 3,* 1 Institute of Physics, University of Rostock, Albert-Einstein-Strasse 23-25, 18059 Rostock, Germany 2 National Science Center Kharkov Institute of Physics and Technology, 61108 Kharkov, Ukraine 3 Institute of Thermal Physics, Ural Branch of the Russian Academy of Sciences, Amundsen Street 107a, 620016 Yekaterinburg, Russia * Correspondence: [email protected]; Tel.: +49-381-498-6889; Fax: +49-381-498-6882 Received: 23 May 2019; Accepted: 5 July 2019; Published: 9 July 2019 Abstract: Thermodynamic aspects of the theory of nucleation are commonly considered employing Gibbs’ theory of interfacial phenomena and its generalizations. Utilizing Gibbs’ theory, the bulk parameters of the critical clusters governing nucleation can be uniquely determined for any metastable state of the ambient phase. As a rule, they turn out in such treatment to be widely similar to the properties of the newly-evolving macroscopic phases. Consequently, the major tool to resolve problems concerning the accuracy of theoretical predictions of nucleation rates and related characteristics of the nucleation process consists of an approach with the introduction of the size or curvature dependence of the surface tension. In the description of crystallization, this quantity has been expressed frequently via changes of entropy (or enthalpy) in crystallization, i.e., via the latent heat of melting or crystallization. Such a correlation between the capillarity phenomena and entropy changes was originally advanced by Stefan considering condensation and evaporation. It is known in the application to crystal nucleation as the Skapski–Turnbull relation. -

2005 Annual Report American Physical Society
1 2005 Annual Report American Physical Society APS 20052 APS OFFICERS 2006 APS OFFICERS PRESIDENT: PRESIDENT: Marvin L. Cohen John J. Hopfield University of California, Berkeley Princeton University PRESIDENT ELECT: PRESIDENT ELECT: John N. Bahcall Leo P. Kadanoff Institue for Advanced Study, Princeton University of Chicago VICE PRESIDENT: VICE PRESIDENT: John J. Hopfield Arthur Bienenstock Princeton University Stanford University PAST PRESIDENT: PAST PRESIDENT: Helen R. Quinn Marvin L. Cohen Stanford University, (SLAC) University of California, Berkeley EXECUTIVE OFFICER: EXECUTIVE OFFICER: Judy R. Franz Judy R. Franz University of Alabama, Huntsville University of Alabama, Huntsville TREASURER: TREASURER: Thomas McIlrath Thomas McIlrath University of Maryland (Emeritus) University of Maryland (Emeritus) EDITOR-IN-CHIEF: EDITOR-IN-CHIEF: Martin Blume Martin Blume Brookhaven National Laboratory (Emeritus) Brookhaven National Laboratory (Emeritus) PHOTO CREDITS: Cover (l-r): 1Diffraction patterns of a GaN quantum dot particle—UCLA; Spring-8/Riken, Japan; Stanford Synchrotron Radiation Lab, SLAC & UC Davis, Phys. Rev. Lett. 95 085503 (2005) 2TESLA 9-cell 1.3 GHz SRF cavities from ACCEL Corp. in Germany for ILC. (Courtesy Fermilab Visual Media Service 3G0 detector studying strange quarks in the proton—Jefferson Lab 4Sections of a resistive magnet (Florida-Bitter magnet) from NHMFL at Talahassee LETTER FROM THE PRESIDENT APS IN 2005 3 2005 was a very special year for the physics community and the American Physical Society. Declared the World Year of Physics by the United Nations, the year provided a unique opportunity for the international physics community to reach out to the general public while celebrating the centennial of Einstein’s “miraculous year.” The year started with an international Launching Conference in Paris, France that brought together more than 500 students from around the world to interact with leading physicists. -

Thermodynamics and Kinetics of Nucleation in Binary Solutions
1 Thermodynamics and kinetics of nucleation in binary solutions Nikolay V. Alekseechkin Akhiezer Institute for Theoretical Physics, National Science Centre “Kharkov Institute of Physics and Technology”, Akademicheskaya Street 1, Kharkov 61108, Ukraine Email: [email protected] Keywords: Binary solution; phase equilibrium; binary nucleation; diffusion growth; surface layer. A new approach that is a combination of classical thermodynamics and macroscopic kinetics is offered for studying the nucleation kinetics in condensed binary solutions. The theory covers the separation of liquid and solid solutions proceeding along the nucleation mechanism, as well as liquid-solid transformations, e.g., the crystallization of molten alloys. The cases of nucleation of both unary and binary precipitates are considered. Equations of equilibrium for a critical nucleus are derived and then employed in the macroscopic equations of nucleus growth; the steady state nucleation rate is calculated with the use of these equations. The present approach can be applied to the general case of non-ideal solution; the calculations are performed on the model of regular solution within the classical nucleation theory (CNT) approximation implying the bulk properties of a nucleus and constant surface tension. The way of extending the theory beyond the CNT approximation is shown in the framework of the finite- thickness layer method. From equations of equilibrium of a surface layer with coexisting bulk phases, equations for adsorption and the dependences of surface tension on temperature, radius, and composition are derived. Surface effects on the thermodynamics and kinetics of nucleation are discussed. 1. Introduction Binary nucleation covers a wide class of processes of phase transformations which can be divided into three groups: (i) gas-liquid (or solid) transformations, (ii) liquid-gas transformations, and (iii) transformations within a condensed state. -

Heterogeneous Nucleation of Water Vapor on Different Types of Black Carbon Particles
https://doi.org/10.5194/acp-2020-202 Preprint. Discussion started: 21 April 2020 c Author(s) 2020. CC BY 4.0 License. Heterogeneous nucleation of water vapor on different types of black carbon particles Ari Laaksonen1,2, Jussi Malila3, Athanasios Nenes4,5 5 1Finnish Meteorological Institute, 00101 Helsinki, Finland. 2Department of Applied Physics, University of Eastern Finland, 70211 Kuopio, Finland. 3Nano and Molecular Systems Research Unit, 90014 University of Oulu, Finland. 4Laboratory of Atmospheric Processes and their Impacts, School of Architecture, Civil & Environmental Engineering, École 10 Polytechnique Federale de Lausanne, 1015, Lausanne, Switzerland 5 Institute of Chemical Engineering Sciences, Foundation for Research and Technology Hellas (FORTH/ICE-HT), 26504, Patras, Greece Correspondence to: Ari Laaksonen ([email protected]) 15 Abstract: Heterogeneous nucleation of water vapor on insoluble particles affects cloud formation, precipitation, the hydrological cycle and climate. Despite its importance, heterogeneous nucleation remains a poorly understood phenomenon that relies heavily on empirical information for its quantitative description. Here, we examine heterogeneous nucleation of water vapor on and cloud drop activation of different types of soots, both pure black carbon particles, and black carbon particles mixed with secondary organic matter. We show that the recently developed adsorption nucleation theory 20 quantitatively predicts the nucleation of water and droplet formation upon particles of the various soot types. A surprising consequence of this new understanding is that, with sufficient adsorption site density, soot particles can activate into cloud droplets – even when completely lacking any soluble material. 1 https://doi.org/10.5194/acp-2020-202 Preprint. Discussion started: 21 April 2020 c Author(s) 2020. -

Determination of Supercooling Degree, Nucleation and Growth Rates, and Particle Size for Ice Slurry Crystallization in Vacuum
crystals Article Determination of Supercooling Degree, Nucleation and Growth Rates, and Particle Size for Ice Slurry Crystallization in Vacuum Xi Liu, Kunyu Zhuang, Shi Lin, Zheng Zhang and Xuelai Li * School of Chemical Engineering, Fuzhou University, Fuzhou 350116, China; [email protected] (X.L.); [email protected] (K.Z.); [email protected] (S.L.); [email protected] (Z.Z.) * Correspondence: [email protected] Academic Editors: Helmut Cölfen and Mei Pan Received: 7 April 2017; Accepted: 29 April 2017; Published: 5 May 2017 Abstract: Understanding the crystallization behavior of ice slurry under vacuum condition is important to the wide application of the vacuum method. In this study, we first measured the supercooling degree of the initiation of ice slurry formation under different stirring rates, cooling rates and ethylene glycol concentrations. Results indicate that the supercooling crystallization pressure difference increases with increasing cooling rate, while it decreases with increasing ethylene glycol concentration. The stirring rate has little influence on supercooling crystallization pressure difference. Second, the crystallization kinetics of ice crystals was conducted through batch cooling crystallization experiments based on the population balance equation. The equations of nucleation rate and growth rate were established in terms of power law kinetic expressions. Meanwhile, the influences of suspension density, stirring rate and supercooling degree on the process of nucleation and growth were studied. Third, the morphology of ice crystals in ice slurry was obtained using a microscopic observation system. It is found that the effect of stirring rate on ice crystal size is very small and the addition of ethylene glycoleffectively inhibits the growth of ice crystals. -
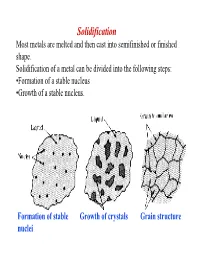
Solidification Most Metals Are Melted and Then Cast Into Semifinished Or Finished Shape
Solidification Most metals are melted and then cast into semifinished or finished shape. Solidification of a metal can be divided into the following steps: •Formation of a stable nucleus •Growth of a stable nucleus. Formation of stable Growth of crystals Grain structure nuclei Polycrystalline Metals In most cases, solidification begins from multiple sites, each of which can produce a different orientation The result is a “polycrystalline” material consisting of many small crystals of “grains” Each grain has the same crystal lattice, but the lattices are misoriented from grain to grain Driving force: solidification ⇒ For the reaction to proceed to the right ∆G AL AS V must be negative. • Writing the free energies of the solid and liquid as: S S S GV = H - TS L L L GV = H - TS ∴∴∴ ∆GV = ∆H - T∆S • At equilibrium, i.e. Tmelt , then the ∆GV = 0 , so we can estimate the melting entropy as: ∆S = ∆H/Tmelt where -∆H is the latent heat (enthalpy) of melting. • Ignore the difference in specific heat between solid and liquid, and we estimate the free energy difference as: T ∆H × ∆T ∆GV ≅ ∆H − ∆H = TMelt TMelt NUCLEATION The two main mechanisms by which nucleation of a solid particles in liquid metal occurs are homogeneous and heterogeneous nucleation. Homogeneous Nucleation Homogeneous nucleation occurs when there are no special objects inside a phase which can cause nucleation. For instance when a pure liquid metal is slowly cooled below its equilibrium freezing temperature to a sufficient degree numerous homogeneous nuclei are created by slow-moving atoms bonding together in a crystalline form. -

The Role of Latent Heat in Heterogeneous Ice Nucleation
EGU21-16164, updated on 02 Oct 2021 https://doi.org/10.5194/egusphere-egu21-16164 EGU General Assembly 2021 © Author(s) 2021. This work is distributed under the Creative Commons Attribution 4.0 License. The role of latent heat in heterogeneous ice nucleation Olli Pakarinen, Cintia Pulido Lamas, Golnaz Roudsari, Bernhard Reischl, and Hanna Vehkamäki University of Helsinki, INAR/Physics, University of Helsinki, Finland ([email protected]) Understanding the way in which ice forms is of great importance to many fields of science. Pure water droplets in the atmosphere can remain in the liquid phase to nearly -40º C. Crystallization of ice in the atmosphere therefore typically occurs in the presence of ice nucleating particles (INPs), such as mineral dust or organic particles, which trigger heterogeneous ice nucleation at clearly higher temperatures. The growth of ice is accompanied by a significant release of latent heat of fusion, which causes supercooled liquid droplets to freeze in two stages [Pruppacher and Klett, 1997]. We are studying these topics by utilizing the monatomic water model [Molinero and Moore, 2009] for unbiased molecular dynamics (MD) simulations, where different surfaces immersed in water are cooled below the melting point over tens of nanoseconds of simulation time and crystallization is followed. With a combination of finite difference calculations and novel moving-thermostat molecular dynamics simulations we show that the release of latent heat from ice growth has a noticeable effect on both the ice growth rate and the initial structure of the forming ice. However, latent heat is found not to be as critically important in controlling immersion nucleation as it is in vapor-to- liquid nucleation [Tanaka et al.2017]. -

Using Supercritical Fluid Technology As a Green Alternative During the Preparation of Drug Delivery Systems
pharmaceutics Review Using Supercritical Fluid Technology as a Green Alternative During the Preparation of Drug Delivery Systems Paroma Chakravarty 1, Amin Famili 2, Karthik Nagapudi 1 and Mohammad A. Al-Sayah 2,* 1 Small Molecule Pharmaceutics, Genentech, Inc. So. San Francisco, CA 94080, USA; [email protected] (P.C.); [email protected] (K.N.) 2 Small Molecule Analytical Chemistry, Genentech, Inc. So. San Francisco, CA 94080, USA; [email protected] * Correspondence: [email protected]; Tel.: +650-467-3810 Received: 3 October 2019; Accepted: 18 November 2019; Published: 25 November 2019 Abstract: Micro- and nano-carrier formulations have been developed as drug delivery systems for active pharmaceutical ingredients (APIs) that suffer from poor physico-chemical, pharmacokinetic, and pharmacodynamic properties. Encapsulating the APIs in such systems can help improve their stability by protecting them from harsh conditions such as light, oxygen, temperature, pH, enzymes, and others. Consequently, the API’s dissolution rate and bioavailability are tremendously improved. Conventional techniques used in the production of these drug carrier formulations have several drawbacks, including thermal and chemical stability of the APIs, excessive use of organic solvents, high residual solvent levels, difficult particle size control and distributions, drug loading-related challenges, and time and energy consumption. This review illustrates how supercritical fluid (SCF) technologies can be superior in controlling the morphology of API particles and in the production of drug carriers due to SCF’s non-toxic, inert, economical, and environmentally friendly properties. The SCF’s advantages, benefits, and various preparation methods are discussed. Drug carrier formulations discussed in this review include microparticles, nanoparticles, polymeric membranes, aerogels, microporous foams, solid lipid nanoparticles, and liposomes. -

Thermodynamics and Kinetics of Binary Nucleation in Ideal-Gas Mixtures
1 Thermodynamics and kinetics of binary nucleation in ideal-gas mixtures Nikolay V. Alekseechkin Akhiezer Institute for Theoretical Physics, National Science Centre “Kharkov Institute of Physics and Technology”, Akademicheskaya Street 1, Kharkov 61108, Ukraine Email: [email protected] Abstract . The nonisothermal single-component theory of droplet nucleation (Alekseechkin, 2014) is extended to binary case; the droplet volume V , composition x , and temperature T are the variables of the theory. An approach based on macroscopic kinetics (in contrast to the standard microscopic model of nucleation operating with the probabilities of monomer attachment and detachment) is developed for the droplet evolution and results in the derived droplet motion equations in the space (V , x,T) - equations for V& ≡ dV / dt , x& , and T& . The work W (V , x,T) of the droplet formation is calculated; it is obtained in the vicinity of the saddle point as a quadratic form with diagonal matrix. Also the problem of generalizing the single-component Kelvin equation for the equilibrium vapor pressure to binary case is solved; it is presented here as a problem of integrability of a Pfaffian equation. The equation for T& is shown to be the first law of thermodynamics for the droplet, which is a consequence of Onsager’s reciprocal relations and the linked-fluxes concept. As an example of ideal solution for demonstrative numerical calculations, the o-xylene-m-xylene system is employed. Both nonisothermal and enrichment effects are shown to exist; the mean steady-state overheat of droplets and their mean steady-state enrichment are calculated with the help of the 3D distribution function.