A CAF40-Binding Motif Facilitates Recruitment of the CCR4-NOT Complex to Mrnas Targeted by Drosophila Roquin
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Towards a Molecular Understanding of Microrna-Mediated Gene Silencing
REVIEWS NON-CODING RNA Towards a molecular understanding of microRNA-mediated gene silencing Stefanie Jonas and Elisa Izaurralde Abstract | MicroRNAs (miRNAs) are a conserved class of small non-coding RNAs that assemble with Argonaute proteins into miRNA-induced silencing complexes (miRISCs) to direct post-transcriptional silencing of complementary mRNA targets. Silencing is accomplished through a combination of translational repression and mRNA destabilization, with the latter contributing to most of the steady-state repression in animal cell cultures. Degradation of the mRNA target is initiated by deadenylation, which is followed by decapping and 5ʹ‑to‑3ʹ exonucleolytic decay. Recent work has enhanced our understanding of the mechanisms of silencing, making it possible to describe in molecular terms a continuum of direct interactions from miRNA target recognition to mRNA deadenylation, decapping and 5ʹ‑to‑3ʹ degradation. Furthermore, an intricate interplay between translational repression and mRNA degradation is emerging. Deadenylation MicroRNAs (miRNAs) are conserved post-transcriptional recruit additional protein partners to mediate silenc- 5,6 Shortening of mRNA poly(A) regulators of gene expression that are integral to ing . Silencing occurs through a combination of tails. In eukaryotes, this almost all known biological processes, including translational repression, deadenylation, decapping and process is catalysed by the cell growth, proliferation and differentiation, as well 5ʹ‑to‑3ʹ mRNA degradation5,6 (FIG. 1). The GW182 pro- consecutive but partially as organismal metabolism and development1. The teins play a central part in this process and are among redundant action of two 5,6 cytoplasmic deadenylase number of miRNAs encoded within the genomes of the most extensively studied AGO partners . -

Inosine in Biology and Disease
G C A T T A C G G C A T genes Review Inosine in Biology and Disease Sundaramoorthy Srinivasan 1, Adrian Gabriel Torres 1 and Lluís Ribas de Pouplana 1,2,* 1 Institute for Research in Biomedicine, Barcelona Institute of Science and Technology, 08028 Barcelona, Catalonia, Spain; [email protected] (S.S.); [email protected] (A.G.T.) 2 Catalan Institution for Research and Advanced Studies, 08010 Barcelona, Catalonia, Spain * Correspondence: [email protected]; Tel.: +34-934034868; Fax: +34-934034870 Abstract: The nucleoside inosine plays an important role in purine biosynthesis, gene translation, and modulation of the fate of RNAs. The editing of adenosine to inosine is a widespread post- transcriptional modification in transfer RNAs (tRNAs) and messenger RNAs (mRNAs). At the wobble position of tRNA anticodons, inosine profoundly modifies codon recognition, while in mRNA, inosines can modify the sequence of the translated polypeptide or modulate the stability, localization, and splicing of transcripts. Inosine is also found in non-coding and exogenous RNAs, where it plays key structural and functional roles. In addition, molecular inosine is an important secondary metabolite in purine metabolism that also acts as a molecular messenger in cell signaling pathways. Here, we review the functional roles of inosine in biology and their connections to human health. Keywords: inosine; deamination; adenosine deaminase acting on RNAs; RNA modification; translation Citation: Srinivasan, S.; Torres, A.G.; Ribas de Pouplana, L. Inosine in 1. Introduction Biology and Disease. Genes 2021, 12, 600. https://doi.org/10.3390/ Inosine was one of the first nucleobase modifications discovered in nucleic acids, genes12040600 having been identified in 1965 as a component of the first sequenced transfer RNA (tRNA), tRNAAla [1]. -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

Novelty Indicator for Enhanced Prioritization of Predicted Gene Ontology Annotations
IEEE/ACM TRANSACTIONS ON COMPUTATIONAL BIOLOGY AND BIOINFORMATICS, VOL. X, NO. X, MONTHXXX 20XX 1 Novelty Indicator for Enhanced Prioritization of Predicted Gene Ontology Annotations Davide Chicco, Fernando Palluzzi, and Marco Masseroli Abstract—Biomolecular controlled annotations have become pivotal in computational biology, because they allow scientists to analyze large amounts of biological data to better understand their test results, and to infer new knowledge. Yet, biomolecular annotation databases are incomplete by definition, like our knowledge of biology, and may contain errors and inconsistent information. In this context, machine-learning algorithms able to predict and prioritize new biomolecular annotations are both effective and efficient, especially if compared with the time-consuming trials of biological validation. To limit the possibility that these techniques predict obvious and trivial high-level features, and to help prioritizing their results, we introduce here a new element that can improve the accuracy and relevance of the results of an annotation prediction and prioritization pipeline. We propose a novelty indicator able to state the level of ”newness” (or ”originality”) of the annotations predicted for a specific gene to Gene Ontology terms, and to help prioritizing the most novel and interesting annotations predicted. We performed a thorough biological functional analysis of the prioritized annotations predicted with high accuracy by using this indicator and our previously proposed prediction algorithms. The relevance -

NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 11-7-2018 Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer Jairo Ramos [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Clinical Epidemiology Commons Recommended Citation Ramos, Jairo, "Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer" (2018). FIU Electronic Theses and Dissertations. 3872. https://digitalcommons.fiu.edu/etd/3872 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida DECIPHER MECHANISMS BY WHICH NUCLEAR RESPIRATORY FACTOR ONE (NRF1) COORDINATES CHANGES IN THE TRANSCRIPTIONAL AND CHROMATIN LANDSCAPE AFFECTING DEVELOPMENT AND PROGRESSION OF INVASIVE BREAST CANCER A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in PUBLIC HEALTH by Jairo Ramos 2018 To: Dean Tomás R. Guilarte Robert Stempel College of Public Health and Social Work This dissertation, Written by Jairo Ramos, and entitled Decipher Mechanisms by Which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer, having been approved in respect to style and intellectual content, is referred to you for judgment. -

Human Social Genomics in the Multi-Ethnic Study of Atherosclerosis
Getting “Under the Skin”: Human Social Genomics in the Multi-Ethnic Study of Atherosclerosis by Kristen Monét Brown A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Epidemiological Science) in the University of Michigan 2017 Doctoral Committee: Professor Ana V. Diez-Roux, Co-Chair, Drexel University Professor Sharon R. Kardia, Co-Chair Professor Bhramar Mukherjee Assistant Professor Belinda Needham Assistant Professor Jennifer A. Smith © Kristen Monét Brown, 2017 [email protected] ORCID iD: 0000-0002-9955-0568 Dedication I dedicate this dissertation to my grandmother, Gertrude Delores Hampton. Nanny, no one wanted to see me become “Dr. Brown” more than you. I know that you are standing over the bannister of heaven smiling and beaming with pride. I love you more than my words could ever fully express. ii Acknowledgements First, I give honor to God, who is the head of my life. Truly, without Him, none of this would be possible. Countless times throughout this doctoral journey I have relied my favorite scripture, “And we know that all things work together for good, to them that love God, to them who are called according to His purpose (Romans 8:28).” Secondly, I acknowledge my parents, James and Marilyn Brown. From an early age, you two instilled in me the value of education and have been my biggest cheerleaders throughout my entire life. I thank you for your unconditional love, encouragement, sacrifices, and support. I would not be here today without you. I truly thank God that out of the all of the people in the world that He could have chosen to be my parents, that He chose the two of you. -

The Metabolites NADP+ and NADPH Are the Targets of the Circadian Protein Nocturnin (Curled)
ARTICLE https://doi.org/10.1038/s41467-019-10125-z OPEN The metabolites NADP+ and NADPH are the targets of the circadian protein Nocturnin (Curled) Michael A. Estrella 1,4, Jin Du 1,4, Li Chen2,3,4, Sneha Rath1, Eliza Prangley1, Alisha Chitrakar1, Tsutomu Aoki1, Paul Schedl1, Joshua Rabinowitz2,3 & Alexei Korennykh1 Nocturnin (NOCT) is a rhythmically expressed protein that regulates metabolism under the control of circadian clock. It has been proposed that NOCT deadenylates and regulates 1234567890():,; metabolic enzyme mRNAs. However, in contrast to other deadenylases, purified NOCT lacks the deadenylase activity. To identify the substrate of NOCT, we conducted a mass spec- trometry screen and report that NOCT specifically and directly converts the dinucleotide NADP+ into NAD+ and NADPH into NADH. Further, we demonstrate that the Drosophila NOCT ortholog, Curled, has the same enzymatic activity. We obtained the 2.7 Å crystal structure of the human NOCT•NADPH complex, which revealed that NOCT recognizes the chemically unique ribose-phosphate backbone of the metabolite, placing the 2′-terminal phosphate productively for removal. We provide evidence for NOCT targeting to mito- chondria and propose that NADP(H) regulation, which takes place at least in part in mito- chondria, establishes the molecular link between circadian clock and metabolism. 1 216 Schultz Laboratory, Department of Molecular Biology, Princeton, NJ 08544, USA. 2 285 Frick Laboratory, Department of Chemistry, Princeton, NJ 08544, USA. 3 Lewis-Sigler Institute for Integrative Genomics, Princeton, NJ 08544, USA. 4These authors contributed equally: Michael A. Estrella, Jin Du, Li Chen. Correspondence and requests for materials should be addressed to J.R. -

Genome-Wide Association Study for Feed Efficiency and Growth Traits in U.S. Beef Cattle Christopher M
Seabury et al. BMC Genomics (2017) 18:386 DOI 10.1186/s12864-017-3754-y RESEARCH ARTICLE Open Access Genome-wide association study for feed efficiency and growth traits in U.S. beef cattle Christopher M. Seabury1*, David L. Oldeschulte1, Mahdi Saatchi2, Jonathan E. Beever3, Jared E. Decker4,5, Yvette A. Halley1, Eric K. Bhattarai1, Maral Molaei1, Harvey C. Freetly6, Stephanie L. Hansen2, Helen Yampara-Iquise4, Kristen A. Johnson7, Monty S. Kerley4, JaeWoo Kim4, Daniel D. Loy2, Elisa Marques8, Holly L. Neibergs7, Robert D. Schnabel4,5, Daniel W. Shike3, Matthew L. Spangler9, Robert L. Weaber10, Dorian J. Garrick2,11 and Jeremy F. Taylor4* Abstract Background: Single nucleotide polymorphism (SNP) arrays for domestic cattle have catalyzed the identification of genetic markers associated with complex traits for inclusion in modern breeding and selection programs. Using actual and imputed Illumina 778K genotypes for 3887 U.S. beef cattle from 3 populations (Angus, Hereford, SimAngus), we performed genome-wide association analyses for feed efficiency and growth traits including average daily gain (ADG), dry matter intake (DMI), mid-test metabolic weight (MMWT), and residual feed intake (RFI), with marker-based heritability estimates produced for all traits and populations. Results: Moderate and/or large-effect QTL were detected for all traits in all populations, as jointly defined by the estimated proportion of variance explained (PVE) by marker effects (PVE ≥ 1.0%) and a nominal P-value threshold (P ≤ 5e-05). Lead SNPs with PVE ≥ 2.0% were considered putative evidence of large-effect QTL (n = 52), whereas those with PVE ≥ 1.0% but < 2.0% were considered putative evidence for moderate-effect QTL (n = 35). -

Differential Regulation of Mrna Fate by the Human Ccr4-Not Complex Is Driven by CDS Composition and Mrna Localisation
bioRxiv preprint doi: https://doi.org/10.1101/2021.03.13.435226; this version posted July 16, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Differential regulation of mRNA fate by the human Ccr4-Not complex is driven by CDS composition and mRNA localisation S.L. Gillen, C.Giacomelli, K. Hodge, S. Zanivan, M. Bushell* & A. Wilczynska* * corresponding authors Abstract Background: Regulation of protein output at the level of translation allows for a rapid adaptation to dynamic changes to the cell’s requirements. This precise control of gene expression is achieved by complex and interlinked biochemical processes that modulate both the protein synthesis rate and stability of each individual mRNA. A major factor coordinating this regulation is the Ccr4-Not complex. Despite playing a role in most stages of the mRNA life cycle, no attempt has been made to take a global integrated view of how the Ccr4-Not complex affects gene expression. Results: This study has taken a comprehensive approach to investigate post-transcriptional regulation mediated by the Ccr4-Not complex assessing steady-state mRNA levels, ribosome position, mRNA stability and protein production transcriptome-wide. Depletion of the scaffold protein CNOT1 results in a global upregulation of mRNA stability and the preferential stabilisation of mRNAs enriched for G/C-ending codons. We also uncover that mRNAs targeted to the ER for their translation have reduced translational efficiency when CNOT1 is depleted, specifically downstream of the signal sequence cleavage site. -

Chromatin Conformation Links Distal Target Genes to CKD Loci
BASIC RESEARCH www.jasn.org Chromatin Conformation Links Distal Target Genes to CKD Loci Maarten M. Brandt,1 Claartje A. Meddens,2,3 Laura Louzao-Martinez,4 Noortje A.M. van den Dungen,5,6 Nico R. Lansu,2,3,6 Edward E.S. Nieuwenhuis,2 Dirk J. Duncker,1 Marianne C. Verhaar,4 Jaap A. Joles,4 Michal Mokry,2,3,6 and Caroline Cheng1,4 1Experimental Cardiology, Department of Cardiology, Thoraxcenter Erasmus University Medical Center, Rotterdam, The Netherlands; and 2Department of Pediatrics, Wilhelmina Children’s Hospital, 3Regenerative Medicine Center Utrecht, Department of Pediatrics, 4Department of Nephrology and Hypertension, Division of Internal Medicine and Dermatology, 5Department of Cardiology, Division Heart and Lungs, and 6Epigenomics Facility, Department of Cardiology, University Medical Center Utrecht, Utrecht, The Netherlands ABSTRACT Genome-wide association studies (GWASs) have identified many genetic risk factors for CKD. However, linking common variants to genes that are causal for CKD etiology remains challenging. By adapting self-transcribing active regulatory region sequencing, we evaluated the effect of genetic variation on DNA regulatory elements (DREs). Variants in linkage with the CKD-associated single-nucleotide polymorphism rs11959928 were shown to affect DRE function, illustrating that genes regulated by DREs colocalizing with CKD-associated variation can be dysregulated and therefore, considered as CKD candidate genes. To identify target genes of these DREs, we used circular chro- mosome conformation capture (4C) sequencing on glomerular endothelial cells and renal tubular epithelial cells. Our 4C analyses revealed interactions of CKD-associated susceptibility regions with the transcriptional start sites of 304 target genes. Overlap with multiple databases confirmed that many of these target genes are involved in kidney homeostasis. -

Gene Expression and Regulation 4
© Jones & Bartlett Learning, LLC. NOT FOR SALE OR DISTRIBUTION Gene expression and regulation 4 David G. Bear Department of Cell Biology and Physiology, University of New Mexico School of Medicine, Albuquerque, NM TRANSLATION OF A MESSENGER RNA (mRNA) isolated from a cell in the salivary gland of an insect Chironomous tentans in this digitally colored trans- mission electron micrograph. Translation is the pro- cess by which ribosomes (blue particles) translocate along the mRNA (pink strand) and read its sequence in three nucleotide units called codons. As the ribo- some moves to each codon on the mRNA, it inserts the appropriate amino acid into the growing protein chain (green filaments). (© Dr. Elena Kiseleva/SPL/ Photo Researchers, Inc.) CHAPTER OUTLINE 4.1 Introduction 4.12 Translation is a three-stage process that decodes an 4.2 Genes are transcription units mRNA to synthesize a protein 4.3 Transcription is a multistep process directed by 4.13 Translation is catalyzed by the ribosome DNA-dependent RNA polymerase 4.14 Translation is guided by a large number of protein 4.4 RNA polymerases are large multisubunit protein factors that regulate the interaction of amino- complexes acylated tRNAs with the ribosome 4.5 Promoters direct the initiation of transcription 4.15 Translation is controlled by the interaction of the 4.6 Activators and repressors regulate transcription 5′ and 3′ ends of the mRNA and by translational initiation repressor proteins 4.7 Transcriptional regulatory circuits control eukaryotic 4.16 Some mRNAs are translated -
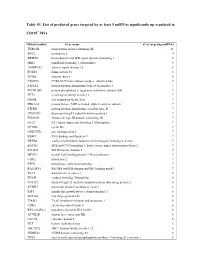
Table S3. List of Predicted Genes Targeted by at Least 5 Mirnas Significantly up Regulated In
Table S3. List of predicted genes targeted by at least 5 miRNAs significantly up regulated in CD105+ MVs. Official symbol Gene name n° of targeting miRNAs TNRC6B trinucleotide repeat containing 6B 11 BNC2 basonuclin 2 9 BRWD1 bromodomain and WD repeat domain containing 1 8 MIB1 mindbomb homolog 1 (Drosophila) 8 ANKRD52 ankyrin repeat domain 52 8 FOXP1 forkhead box P1 8 ITGB8 integrin, beta 8 8 CNOT6L CCR4-NOT transcription complex, subunit 6-like 8 PTP4A1 protein tyrosine phosphatase type IVA, member 1 7 PPP1R15B protein phosphatase 1, regulatory (inhibitor) subunit 15B 7 TET3 tet oncogene family member 3 7 GMFB glia maturation factor, beta 7 PRKAA2 protein kinase, AMP-activated, alpha 2 catalytic subunit 7 PTPRD protein tyrosine phosphatase, receptor type, D 7 TP53INP1 tumor protein p53 inducible nuclear protein 1 7 FNDC3B fibronectin type III domain containing 3B 7 FAT3 FAT tumor suppressor homolog 3 (Drosophila) 7 CCND2 cyclin D2 7 ONECUT2 one cut homeobox 2 7 RBM9 RNA binding motif protein 9 7 ERBB4 v-erb-a erythroblastic leukemia viral oncogene homolog 4 (avian) 7 BACH2 BTB and CNC homology 1, basic leucine zipper transcription factor 2 7 SMAD4 SMAD family member 4 7 MECP2 methyl CpG binding protein 2 (Rett syndrome) 7 UBN2 ubinuclein 2 7 PTEN phosphatase and tensin homolog 7 RALGPS1 Ral GEF with PH domain and SH3 binding motif 1 7 DLC1 deleted in liver cancer 1 6 ENAH enabled homolog (Drosophila) 6 NUFIP2 nuclear fragile X mental retardation protein interacting protein 2 6 KCMF1 potassium channel modulatory factor 1 6 IGF1 insulin-like growth factor 1 (somatomedin C) 6 ZNF148 zinc finger protein 148 6 TIAM1 T-cell lymphoma invasion and metastasis 1 6 CDK6 cyclin-dependent kinase 6 6 RP5-1022P6.2 hypothetical protein KIAA1434 6 ACVR2B activin A receptor, type IIB 6 CLCN5 chloride channel 5 6 HLF hepatic leukemia factor 6 TBC1D12 TBC1 domain family, member 12 6 FRMD4A FERM domain containing 4A 6 NUS1 nuclear undecaprenyl pyrophosphate synthase 1 homolog (S.