Bacterial Diversity Analysis of Yumthang Hot Spring, North Sikkim
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Changes in Bacterioplankton Communities Resulting from Direct and Indirect Interactions with Trace Metal Gradients in an Urbaniz
Changes in Bacterioplankton Communities Resulting From Direct and Indirect Interactions With Trace Metal Gradients in an Urbanized Marine Coastal Area Clément Coclet, Cédric Garnier, Gaël Durrieu, Dario Omanović, Sébastien d’Onofrio, Christophe Le Poupon, Jean-Ulrich Mullot, Jean-François Briand, Benjamin Misson To cite this version: Clément Coclet, Cédric Garnier, Gaël Durrieu, Dario Omanović, Sébastien d’Onofrio, et al.. Changes in Bacterioplankton Communities Resulting From Direct and Indirect Interactions With Trace Metal Gradients in an Urbanized Marine Coastal Area. Frontiers in Microbiology, Frontiers Media, 2019, 10, 10.3389/fmicb.2019.00257. hal-02049263 HAL Id: hal-02049263 https://hal.archives-ouvertes.fr/hal-02049263 Submitted on 26 Feb 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. fmicb-10-00257 February 20, 2019 Time: 17:14 # 1 ORIGINAL RESEARCH published: 22 February 2019 doi: 10.3389/fmicb.2019.00257 Changes in Bacterioplankton Communities Resulting From Direct and Indirect Interactions With Trace Metal Gradients in an Urbanized -

Thiobacillus Denitrificans
Nitrate-Dependent, Neutral pH Bioleaching of Ni from an Ultramafic Concentrate by Han Zhou A thesis submitted in conformity with the requirements for the degree of Master of Applied Science Chemical Engineering and Applied Chemistry University of Toronto © Copyright by Han Zhou 2014 ii Nitrate-Dependent, Neutral pH Bioleaching of Ni from an Ultramafic Concentrate Han Zhou Master of Applied Science Chemical Engineering and Applied Chemistry University of Toronto 2014 Abstract This study explores the possibility of utilizing bioleaching techniques for nickel extraction from a mixed sulfide ore deposit with high magnesium content. Due to the ultramafic nature of this material, well-studied bioleaching technologies, which rely on acidophilic bacteria, will lead to undesirable processing conditions. This is the first work that incorporates nitrate-dependent bacteria under pH 6.5 environments for bioleaching of base metals. Experiments with both defined bacterial strains and indigenous mixed bacterial cultures were conducted with nitrate as the electron acceptor and sulfide minerals as electron donors in a series of microcosm studies. Nitrate consumption, sulfate production, and Ni released into the aqueous phase were used to track the extent of oxidative sulfide mineral dissolution; taxonomic identification of the mixed culture community was performed using 16S rRNA gene sequencing. Nitrate-dependent microcosms that contained indigenous sulfur- and/or iron-oxidizing microorganisms were cultured, characterized, and provided a proof-of-concept basis for further bioleaching studies. iii Acknowledgments I would like to extend my most sincere gratitude toward both of my supervisors Dr. Vladimiros Papangelakis and Dr. Elizabeth Edwards. This work could not have been completed without your brilliant and patient guidance. -

Downloaded 13 April 2017); Using Diamond
bioRxiv preprint doi: https://doi.org/10.1101/347021; this version posted June 14, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 2 3 4 5 Re-evaluating the salty divide: phylogenetic specificity of 6 transitions between marine and freshwater systems 7 8 9 10 Sara F. Pavera, Daniel J. Muratorea, Ryan J. Newtonb, Maureen L. Colemana# 11 a 12 Department of the Geophysical Sciences, University of Chicago, Chicago, Illinois, USA 13 b School of Freshwater Sciences, University of Wisconsin Milwaukee, Milwaukee, Wisconsin, USA 14 15 Running title: Marine-freshwater phylogenetic specificity 16 17 #Address correspondence to Maureen Coleman, [email protected] 18 bioRxiv preprint doi: https://doi.org/10.1101/347021; this version posted June 14, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 19 Abstract 20 Marine and freshwater microbial communities are phylogenetically distinct and transitions 21 between habitat types are thought to be infrequent. We compared the phylogenetic diversity of 22 marine and freshwater microorganisms and identified specific lineages exhibiting notably high or 23 low similarity between marine and freshwater ecosystems using a meta-analysis of 16S rRNA 24 gene tag-sequencing datasets. As expected, marine and freshwater microbial communities 25 differed in the relative abundance of major phyla and contained habitat-specific lineages; at the 26 same time, however, many shared taxa were observed in both environments. 27 Betaproteobacteria and Alphaproteobacteria sequences had the highest similarity between 28 marine and freshwater sample pairs. -

Evaluating the Biogeochemistry and Microbial Function in the Athabasca Oil Sands Region: Understanding Natural Baselines for Reclamation End-Points
University of Windsor Scholarship at UWindsor Electronic Theses and Dissertations Theses, Dissertations, and Major Papers 2019 Evaluating the biogeochemistry and microbial function in the Athabasca Oil Sands region: Understanding natural baselines for reclamation end-points Thomas Reid University of Windsor Follow this and additional works at: https://scholar.uwindsor.ca/etd Recommended Citation Reid, Thomas, "Evaluating the biogeochemistry and microbial function in the Athabasca Oil Sands region: Understanding natural baselines for reclamation end-points" (2019). Electronic Theses and Dissertations. 7732. https://scholar.uwindsor.ca/etd/7732 This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email ([email protected]) or by telephone at 519-253-3000ext. 3208. Evaluating the biogeochemistry and microbial function in the Athabasca Oil Sands -

Supplementary Information for Microbial Electrochemical Systems Outperform Fixed-Bed Biofilters for Cleaning-Up Urban Wastewater
Electronic Supplementary Material (ESI) for Environmental Science: Water Research & Technology. This journal is © The Royal Society of Chemistry 2016 Supplementary information for Microbial Electrochemical Systems outperform fixed-bed biofilters for cleaning-up urban wastewater AUTHORS: Arantxa Aguirre-Sierraa, Tristano Bacchetti De Gregorisb, Antonio Berná, Juan José Salasc, Carlos Aragónc, Abraham Esteve-Núñezab* Fig.1S Total nitrogen (A), ammonia (B) and nitrate (C) influent and effluent average values of the coke and the gravel biofilters. Error bars represent 95% confidence interval. Fig. 2S Influent and effluent COD (A) and BOD5 (B) average values of the hybrid biofilter and the hybrid polarized biofilter. Error bars represent 95% confidence interval. Fig. 3S Redox potential measured in the coke and the gravel biofilters Fig. 4S Rarefaction curves calculated for each sample based on the OTU computations. Fig. 5S Correspondence analysis biplot of classes’ distribution from pyrosequencing analysis. Fig. 6S. Relative abundance of classes of the category ‘other’ at class level. Table 1S Influent pre-treated wastewater and effluents characteristics. Averages ± SD HRT (d) 4.0 3.4 1.7 0.8 0.5 Influent COD (mg L-1) 246 ± 114 330 ± 107 457 ± 92 318 ± 143 393 ± 101 -1 BOD5 (mg L ) 136 ± 86 235 ± 36 268 ± 81 176 ± 127 213 ± 112 TN (mg L-1) 45.0 ± 17.4 60.6 ± 7.5 57.7 ± 3.9 43.7 ± 16.5 54.8 ± 10.1 -1 NH4-N (mg L ) 32.7 ± 18.7 51.6 ± 6.5 49.0 ± 2.3 36.6 ± 15.9 47.0 ± 8.8 -1 NO3-N (mg L ) 2.3 ± 3.6 1.0 ± 1.6 0.8 ± 0.6 1.5 ± 2.0 0.9 ± 0.6 TP (mg -

An Oligotrophic Deep-Subsurface Community Dependent on Syntrophy
An oligotrophic deep-subsurface community PNAS PLUS dependent on syntrophy is dominated by sulfur-driven autotrophic denitrifiers Maggie C. Y. Laua,1, Thomas L. Kieftb, Olukayode Kuloyoc,2, Borja Linage-Alvarezc,3, Esta van Heerdenc, Melody R. Lindsaya,4, Cara Magnaboscoa,5, Wei Wangd, Jessica B. Wigginsd, Ling Guod, David H. Perlmane,6, Saw Kyine, Henry H. Shwee, Rachel L. Harrisa, Youmi Ohf,7, Min Joo Yig, Roland Purtscherth, Greg F. Slateri, Shuhei Onoj, Siwen Weik, Long Lik,l, Barbara Sherwood Lollarl, and Tullis C. Onstotta aDepartment of Geosciences, Princeton University, Princeton, NJ 08544; bDepartment of Biology, New Mexico Institute of Mining and Technology, Socorro, NM 87801; cDepartment of Microbial, Biochemical, and Food Biotechnology, University of the Free State, Bloemfontein 9301, South Africa; dHigh Throughput Sequencing and Microarray Facility, Lewis–Sigler Institute for Integrative Genomics, Princeton University, NJ 08544; eProteomics and Mass Spectrometry Core, Department of Molecular Biology, Princeton University, NJ 08544; fAtmospheric and Oceanic Sciences, Princeton University, Princeton, NJ 08544; gDepartment of Ecology and Evolutionary Biology, Princeton University, Princeton, NJ 08544; hClimate and Environmental Physics, Physics Institute, University of Bern, 3012 Bern, Switzerland; iSchool of Geography and Earth Sciences, McMaster University, Hamilton, ON, Canada L8S 4K1; jDepartment of Earth, Atmospheric, and Planetary Sciences, Massachusetts Institute of Technology, Cambridge, MA 02139; kDepartment of Earth and Atmospheric Sciences, University of Alberta, Edmonton, AB, Canada T6G 2E3; and lDepartment of Earth Sciences, University of Toronto, Toronto, ON, Canada M5S 3B1 Edited by David M. Karl, University of Hawaii, Honolulu, HI, and approved October 26, 2016 (received for review August 10, 2016) Subsurface lithoautotrophic microbial ecosystems (SLiMEs) under reasonable to hypothesize that subsurface inhabitants with diverse oligotrophic conditions are typically supported by H2. -

Microbial Community Composition and Activities in Wet Flue Gas
MICROBIAL COMMUNITY COMPOSITION AND ACTIVITIES IN WET FLUE GAS DESULFURIZATION SYSTEMS A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Gregory D. Martin May, 2017 MICROBIAL COMMUNITY COMPOSITION AND ACTIVITIES IN WET FLUE GAS DESULFURIZATION SYSTEMS Gregory D. Martin Thesis Approved: Accepted: ______________________________________ _______________________________________ Advisor Dean of the College Dr. John M. Senko Dr. John Green ______________________________________ _______________________________________ Faculty Reader Dean of the Graduate School Dr. Teresa J. Cutright Dr. Chand Midha ______________________________________ _______________________________________ Faculty Reader Date Dr. Hazel A. Barton ______________________________________ Department Chair Dr. Stephen C. Weeks ii ABSTRACT This project was conducted to characterize microbial communities and slurry in wet flue gas desulfurization (wFGD) units at coal burning power plants. An additional objective of this research was to ascertain microbial activity and the potential for microbial mercury metabolism. Coal fired power plants in the U.S. alone are responsible for emitting over 50 tons per year of Hg0 into the atmosphere. A consequence of this microbially produced MeHg is increased toxicity with distance in food webs, eventually reaching humans where it can damage nervous systems and impair fetal development. Therefore, Hg bioaccumulation as a direct result of increased anthropogenic Hg0 emissions is a global concern. To address the chemistry and microbial activities of wFGD slurry, I determined the physiochemisty of three wFGD systems. I then quantified the activity of microorganisms in the wFGD slurry using live/dead cell counts, respirometry experiments monitoring O2 consumption over time, a Hg reduction experiment monitoring total Hg loss over time, and total RNA sequencing reads. -

Manuscript III
Manuscript III 60 Manuscript Initial characterization of ParI, an orphan C5-DNA methyltransferase from Psychrobacter arcticus 273-4 Miriam Grgic1, Gro Elin Kjæreng Bjerga2, Adele Kim Williamson1, Bjørn Altermark1 and Ingar Leiros1 1Department of chemistry, University of Tromsø, The Arctic University of Norway, Tromsø, Norway 2 University of Bergen, Bergen, Norway 1 Abstract Background DNA methyltransferase are important enzymes in most living organisms, from single cell bacteria to eukaryotes. In prokaryotes, most DNA methyltransferases are members of the bacterial type II restriction modification system: The DNA methyltransferases methylates host DNA, thereby protecting it from cleavage by the restriction endonucleases that recognize the same sequence. In addition to being part of restriction modification systems, DNA methyltransferases can act as orphan enzymes having important role in controlling gene expression, DNA replication, cell cycle and DNA post replicative mismatch repair. In higher eukaryotes, DNA methylation is involved in the regulation of gene expression, maintenance of genome integrity, parental imprinting, chromatin condensation as well as being involved in development of diseases and disorders. DNA-MTases have several potential applications in biotechnology for example in targeted gene silencing and in DNA-protein interaction studies Results According to bioinformatical analyses, the parI gene from Psychrobacter arcticus 273-4, encoding a cytosine specific DNA methyltransferase, is likely of phage origin. In this work, ParI was expressed and purified in a McrBC negative E. coli strain in fusion with a hexahistidine affinity tag and maltose binding protein solubility protein. The obtained recombinant ParI was monomeric with a molecular mass estimated to 54 kDa. The melting temperature of the protein was determined to be 53 ºC in ThermoFlour assay and 54 ºC in differential scanning calorimetry, with no secondary structures detectable at 65 ºC. -

Fish Bacterial Flora Identification Via Rapid Cellular Fatty Acid Analysis
Fish bacterial flora identification via rapid cellular fatty acid analysis Item Type Thesis Authors Morey, Amit Download date 09/10/2021 08:41:29 Link to Item http://hdl.handle.net/11122/4939 FISH BACTERIAL FLORA IDENTIFICATION VIA RAPID CELLULAR FATTY ACID ANALYSIS By Amit Morey /V RECOMMENDED: $ Advisory Committe/ Chair < r Head, Interdisciplinary iProgram in Seafood Science and Nutrition /-■ x ? APPROVED: Dean, SchooLof Fisheries and Ocfcan Sciences de3n of the Graduate School Date FISH BACTERIAL FLORA IDENTIFICATION VIA RAPID CELLULAR FATTY ACID ANALYSIS A THESIS Presented to the Faculty of the University of Alaska Fairbanks in Partial Fulfillment of the Requirements for the Degree of MASTER OF SCIENCE By Amit Morey, M.F.Sc. Fairbanks, Alaska h r A Q t ■ ^% 0 /v AlA s ((0 August 2007 ^>c0^b Abstract Seafood quality can be assessed by determining the bacterial load and flora composition, although classical taxonomic methods are time-consuming and subjective to interpretation bias. A two-prong approach was used to assess a commercially available microbial identification system: confirmation of known cultures and fish spoilage experiments to isolate unknowns for identification. Bacterial isolates from the Fishery Industrial Technology Center Culture Collection (FITCCC) and the American Type Culture Collection (ATCC) were used to test the identification ability of the Sherlock Microbial Identification System (MIS). Twelve ATCC and 21 FITCCC strains were identified to species with the exception of Pseudomonas fluorescens and P. putida which could not be distinguished by cellular fatty acid analysis. The bacterial flora changes that occurred in iced Alaska pink salmon ( Oncorhynchus gorbuscha) were determined by the rapid method. -
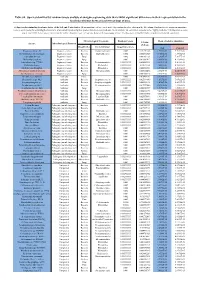
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

Characterization and Biological Activity of Bacterial Glycoconjugates in Cold Adaptation
UNIVERSITA’ DEGLI STUDI DI NAPOLI FEDERICO II Chemical Science XXVIII Cycle Characterization and biological activity of bacterial glycoconjugates in cold adaptation. Angela Casillo Tutor: Prof. Maria Michela Corsaro Supervisor: Prof. Angela Amoresano 1 Summary Abbreviation 6 Abstract 8 Introduction Chapter I: Microorganisms at the limits of life 17 1.1 Psychrophiles 1.2 Molecular and physiological adaptation 1.3 Industrial and biotechnological applications Chapter II: Gram-negative bacteria 25 2.1 Gram-negative cell membrane: Lipopolysaccharide 2.2 Bacterial extracellular polysaccharides (CPSs and EPSs) 2.3 Biofilm 2.4 Polyhydroxyalkanoates (PHAs Chapter III: Methodology 36 3.1 Extraction and purification of LPS 3.2 Chemical analysis and reactions on LPS 3.2.1 Lipid A structure determination 3.2.2 Core region determination 3.3 Chromatography in the study of oligo/polysaccharides 3.4 Mass spectrometry of oligo/polysaccharides 3.5 Nuclear Magnetic Resonance (NMR) Results Colwellia psychrerythraea strain 34H Chapter IV: C. psychrerythraea grown at 4°C 47 4.1 Lipopolysaccharide and lipid A structures 4.1.1Isolation and purification of lipid A 4.1.2 ESI FT-ICR mass spectrometric analysis of lipid A 4.1.3 Discussion 4.2 Capsular polysaccharide (CPS) 4.2.1 Isolation and purification of CPS 4.2.2 NMR analysis of purified CPS 4.2.3 Three-dimensional structure characterization 4.2.4 Ice recrystallization inhibition assay 4.2.5 Discussion 4.3 Extracellular polysaccharide (EPS) 4.3.1 Isolation and purification of EPS 4.3.2 NMR analysis of purified EPS 4.3.3 Three-Dimensional Structure Characterization 4.3.4 Ice Recrystallization Inhibition assay 4.3.5 Discussion 4.4 Mannan polysaccharide 4.4.1 Isolation and purification of Mannan polysaccharide 4.4.2 NMR analysis 4.4.3 Ice Recrystallization Inhibition assay 4.4.4 Discussion 4.5 Polyhydroxyalkanoates (PHA) 4.5.1 Discussion Conclusion Chapter V: C. -

Marine Drugs
Mar. Drugs 2015, 13, 4539-4555; doi:10.3390/md13074539 OPEN ACCESS marine drugs ISSN 1660-3397 www.mdpi.com/journal/marinedrugs Article Structural Investigation of the Oligosaccharide Portion Isolated from the Lipooligosaccharide of the Permafrost Psychrophile Psychrobacter arcticus 273-4 Angela Casillo 1, Ermenegilda Parrilli 1, Sannino Filomena 1,2, Buko Lindner 3, Rosa Lanzetta 1, Michelangelo Parrilli 4, Maria Luisa Tutino 1 and Maria Michela Corsaro 1,* 1 Dipartimento di Scienze Chimiche, Università degli Studi di Napoli Federico II, Complesso Universitario Monte S. Angelo, Via Cintia 4, Napoli 80126, Italy; E-Mails: [email protected] (A.C.); [email protected] (E.P.); [email protected] (S.F.); [email protected] (R.L.); [email protected] (M.L.T.) 2 Institute of Protein Biochemistry, CNR, Via Pietro Castellino 111, Napoli 80131, Italy 3 Division of Bioanalytical Chemistry, Research Center Borstel, Leibniz-Center for Medicine and Biosciences, Parkallee 10, BorstelD-23845, Germany; E-Mail: [email protected] 4 Dipartimento di Biologia, Università degli Studi di Napoli Federico II, Complesso Universitario Monte S. Angelo, Via Cintia 4, Napoli 80126, Italy; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +39-081-674149; Fax: +39-081-674393. Academic Editor: Antonio Trincone Received: 22 June 2015 / Accepted: 14 July 2015 /Published: 22 July 2015 Abstract: Psychrophilic microorganisms have successfully colonized all permanently cold environments from the deep sea to mountain and polar regions. The ability of an organism to survive and grow in cryoenviroments depends on a number of adaptive strategies aimed at maintaining vital cellular functions at subzero temperatures, which include the structural modifications of the membrane.