2021.03.19.436079V1.Full.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mutated SF3B1 Is Associated with Transcript Isoform Changes of The
bioRxiv preprint doi: https://doi.org/10.1101/000992; this version posted July 13, 2014. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Reyes et al. RESEARCH Mutated SF3B1 is associated with transcript isoform changes of the genes UQCC and RPL31 both in CLLs and uveal melanomas Alejandro Reyes1, Carolin Blume2, Vincent Pelechano1, Petra Jakob1, Lars M Steinmetz1,3, Thorsten Zenz2,4 and Wolfgang Huber1* *Correspondence: [email protected] 1European Molecular Biology Abstract Laboratory, Genome Biology Unit, 69117, Heidelberg Germany Background: Genome sequencing studies of chronic lympoid leukemia (CLL) Full list of author information is have provided a comprehensive overview of recurrent somatic mutations in coding available at the end of the article genes. One of the most intriguing discoveries has been the prevalence of mutations in the HEAT-repeat domain of the splicing factor SF3B1. A frequently observed variant is predicted to cause the substitution of a lysine with a glutamic acid at position 700 of the protein (K700E). However, the molecular consequences of the mutations are largely unknown. Results: To start exploring this question, we sequenced the transcriptomes of six samples: four samples of CLL tumour cells, of which two contained the K700E mutation in SF3B1, and CD19 positive cells from two healthy donors. We identified 41 genes that showed differential usage of exons statistically associated with the mutated status of SF3B1 (false discovery rate of 10%). -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Anti-Phospho-SF3B1 (Sap155) (Ser129) Pab
PD043 Page 1 For Research Use Only. Not for use in diagnostic procedures. Anti-Phospho-SF3B1 (Sap155) (Ser129) pAb CODE No. PD043 CLONALITY Polyclonal ISOTYPE Rabbit Ig, affinity purified QUANTITY 100 L SOURCE Purified IgG from rabbit serum IMMUNOGEN KLH conjugated synthetic peptide, RTMII(pS)PERL (corresponding to amino acid residues 124-133 of human SF3B1) FORMURATION PBS containing 50% Glycerol (pH 7.2). No preservative is contained. STORAGE This antibody solution is stable for one year from the date of purchase when stored at -20°C. APPLICATION-CONFIRMED Western blotting 1:1000 for chemiluminescence detection system SPECIES CROSS REACTIVITY on WB Species Human Mouse Rat Hamster Cell HeLa, Raji NIH/3T3, WR19L Not tested Not tested Reactivity Entrez Gene ID 23451 (Human), 81898 (Mouse) REFERENCES 1) Wang, C., et al., Genes Dev. 12, 1409-1414 (1998) 2) Shi, Y., et al., Mol Cell 23, 819-829 (2006) 3) Tanuma, N., et al., J. Biol. Chem., 283, 35805-35814 (2008) 4) Yoshida, K., et al., Nature 478, 64-69 (2011) 5) Rossi, D., et al., Blood 118, 6904-6908 (2011) 6) Quesada, V., et al., Nat Genet 44, 47-52 (2011) 7) Wang, L., et al., N Engl J Med. 365, 2497-2506 (2011) 8) Biankin, A. V., et al., Nature 491, 399-405 (2012) For more information, please visit our web site http://ruo.mbl.co.jp/ MEDICAL & BIOLOGICAL LABORATORIES CO., LTD. URL http://ruo.mbl.co.jp/ e-mail [email protected], TEL 052-238-1904 PD043 Page 2 P a g RELATED PRODUCTS e PD043 Anti-Phospho-SF3B1 (Sap155) (Ser129) pAb D138-3 Anti-Sap155 (SF3B12) mAb D221-3 Anti-Sap155 (SF3B1) -

Monoclonal B-Cell Lymphocytosis Is Characterized by Mutations in CLL Putative Driver Genes and Clonal Heterogeneity Many Years Before Disease Progression
Leukemia (2014) 28, 2395–2424 © 2014 Macmillan Publishers Limited All rights reserved 0887-6924/14 www.nature.com/leu LETTERS TO THE EDITOR Monoclonal B-cell lymphocytosis is characterized by mutations in CLL putative driver genes and clonal heterogeneity many years before disease progression Leukemia (2014) 28, 2395–2398; doi:10.1038/leu.2014.226 (Beckton Dickinson) and data analyzed using Cell Quest software. On the basis of FACS (fluorescence-activated cell sorting) analysis, we observed after enrichment an average of 91% of CD19+ cells Monoclonal B-cell lymphocytosis (MBL) is defined as an asympto- (range 76–99%) and 91% of the CD19+ fraction were CD19+/CD5+ matic expansion of clonal B cells with less than 5 × 109/L cells in the cells (range 66–99%). We used the values of the CD19+/CD5+ peripheral blood and without other manifestations of chronic fraction to calculate the leukemic B-cell fraction and reduce any lymphocytic leukemia (CLL; for example, lymphadenopathy, cyto- significant contamination of non-clonal B cells in each biopsy. DNA penias, constitutional symptoms).1 Approximately 1% of the MBL was extracted from the clonal B cells and non-clonal (that is, T cells) cohort develops CLL per year. Evidence suggests that nearly all CLL cells using the Gentra Puregene Cell Kit (Qiagen, Hilden, Germany). 2 fi fi cases are preceded by an MBL state. Our understanding of the Extracted DNAs were ngerprinted to con rm the relationship genetic basis, clonal architecture and evolution in CLL pathogenesis between samples of the same MBL individual and to rule out sample has undergone significant improvements in the last few years.3–8 In cross-contamination between individuals. -

SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression
Published OnlineFirst August 20, 2019; DOI: 10.1158/0008-5472.CAN-18-3965 Cancer Molecular Cell Biology Research SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression Norihiko Kawamura1,2, Keisuke Nimura1, Kotaro Saga1, Airi Ishibashi1, Koji Kitamura1,3, Hiromichi Nagano1, Yusuke Yoshikawa4, Kyoso Ishida1,5, Norio Nonomura2, Mitsuhiro Arisawa4, Jun Luo6, and Yasufumi Kaneda1 Abstract Androgen receptor splice variant-7 (AR-V7) is a General RNA splicing SF3B2 complex-mediated alternative RNA splicing constitutively active AR variant implicated in U2 castration-resistant prostate cancers. Here, we show U2 snRNA that the RNA splicing factor SF3B2, identified by 3’ 3’ in silico and CRISPR/Cas9 analyses, is a critical 5’ 3’ splice site 5’ SF3B7 AR-V7 5’ A U2AF2 AGA Exon ? determinant of expression and is correlated SF3B6(p14) SF3B4 SF3B1 SF3B4 SF3B1 with aggressive cancer phenotypes. Transcriptome SF3B5 SF3B2 SF3B3 SF3B2 SF3B3 and PAR-CLIP analyses revealed that SF3B2 con- SF3A3 SF3B2 complex SF3A3 SF3A1 SF3A1 SF3b complex trols the splicing of target genes, including AR, to AR pre-mRNA drive aggressive phenotypes. SF3B2-mediated CE3 aggressive phenotypes in vivo were reversed by AR-V7 mRNA AR mRNA AR-V7 knockout. Pladienolide B, an inhibitor of CE3 a splicing modulator of the SF3b complex, sup- Drive malignancy pressed the growth of tumors addicted to high While the SF3b complex is critical for general RNA splicing, SF3B2 promotes inclusion of the target exon through recognizing a specific RNA motif. SF3B2 expression. These findings support the idea © 2019 American Association for Cancer Research that alteration of the splicing pattern by high SF3B2 expression is one mechanism underlying prostate cancer progression and therapeutic resistance. -

Global Proteomic Analysis Deciphers the Mechanism of Action of Plant
www.nature.com/scientificreports OPEN Global proteomic analysis deciphers the mechanism of action of plant derived oleic acid against Candida albicans virulence and bioflm formation Subramanian Muthamil1,2, Krishnan Ganesh Prasath1,2, Arumugam Priya1, Pitchai Precilla1 & Shunmugiah Karutha Pandian1* Candida albicans is a commensal fungus in humans, mostly found on the mucosal surfaces of the mouth, gut, vagina and skin. Incidence of ever increasing invasive candidiasis in immunocompromised patients, alarming occurrence of antifungal resistance and insufcient diagnostic methods demand more focused research into C. albicans pathogenicity. Consequently, in the present study, oleic acid from Murraya koenigii was shown to have the efcacy to inhibit bioflm formation and virulence of Candida spp. Results of in vitro virulence assays and gene expression analysis, impelled to study the protein targets which are involved in the molecular pathways of C. albicans pathogenicity. Proteomic studies of diferentially expressed proteins reveals that oleic acid induces oxidative stress responses and mainly targets the proteins involved in glucose metabolism, ergosterol biosynthesis, lipase production, iron homeostasis and amino acid biosynthesis. The current study emphasizes anti-virulent potential of oleic acid which can be used as a therapeutic agent to treat Candida infections. Candida is a genus of yeast with remarkable phenotypic characteristics and found as a commensal fungus in humans. Candida species are most commonly present in the genital tracts and other membrane tracts such as mucosal oral cavity, respiratory tract, gastrointestinal tract etc1. Although over hundred Candida species belong to this genus, C. albicans is responsible for the majority of Candida infection. Other medically important Candida species are Candida glabrata, Candida tropicalis, Candida dubliniensis, and Candida parapsilosis2. -

Flexible, Unbiased Analysis of Biological Characteristics Associated with Genomic Regions
bioRxiv preprint doi: https://doi.org/10.1101/279612; this version posted March 22, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. BioFeatureFinder: Flexible, unbiased analysis of biological characteristics associated with genomic regions Felipe E. Ciamponi 1,2,3; Michael T. Lovci 2; Pedro R. S. Cruz 1,2; Katlin B. Massirer *,1,2 1. Structural Genomics Consortium - SGC, University of Campinas, SP, Brazil. 2. Center for Molecular Biology and Genetic Engineering - CBMEG, University of Campinas, Campinas, SP, Brazil. 3. Graduate program in Genetics and Molecular Biology, PGGBM, University of Campinas, Campinas, SP, Brazil. *Corresponding author: [email protected] Mailing address: Center for Molecular Biology and Genetic Engineering - CBMEG, University of Campinas, Campinas, SP, Brazil. Av Candido Rondo, 400 Cidade Universitária CEP 13083-875, Campinas, SP Phone: 55-19-98121-937 bioRxiv preprint doi: https://doi.org/10.1101/279612; this version posted March 22, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Abstract BioFeatureFinder is a novel algorithm which allows analyses of many biological genomic landmarks (including alternatively spliced exons, DNA/RNA- binding protein binding sites, and gene/transcript functional elements, nucleotide content, conservation, k-mers, secondary structure) to identify distinguishing features. -

Full-Length Transcript Characterization of SF3B1 Mutation in Chronic Lymphocytic Leukemia Reveals Downregulation of Retained Introns
ARTICLE https://doi.org/10.1038/s41467-020-15171-6 OPEN Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns Alison D. Tang 1, Cameron M. Soulette 2, Marijke J. van Baren1, Kevyn Hart1, Eva Hrabeta-Robinson1, ✉ Catherine J. Wu 3,4,5 & Angela N. Brooks 1 SF3B1 1234567890():,; While splicing changes caused by somatic mutations in are known, identifying full- length isoform changes may better elucidate the functional consequences of these mutations. We report nanopore sequencing of full-length cDNA from CLL samples with and without SF3B1 mutation, as well as normal B cell samples, giving a total of 149 million pass reads. We present FLAIR (Full-Length Alternative Isoform analysis of RNA), a computational workflow to identify high-confidence transcripts, perform differential splicing event analysis, and dif- ferential isoform analysis. Using nanopore reads, we demonstrate differential 3’ splice site changes associated with SF3B1 mutation, agreeing with previous studies. We also observe a strong downregulation of intron retention events associated with SF3B1 mutation. Full-length transcript analysis links multiple alternative splicing events together and allows for better estimates of the abundance of productive versus unproductive isoforms. Our work demon- strates the potential utility of nanopore sequencing for cancer and splicing research. 1 Department of Biomolecular Engineering, University of California, Santa Cruz, CA 95062, USA. 2 Department of Molecular Cell & Developmental Biology, University of California, Santa Cruz, CA 95062, USA. 3 Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA 02215, USA. 4 Broad Institiute of Harvard and MIT, Cambridge, MA, USA. -

A Peripheral Blood Gene Expression Signature to Diagnose Subclinical Acute Rejection
CLINICAL RESEARCH www.jasn.org A Peripheral Blood Gene Expression Signature to Diagnose Subclinical Acute Rejection Weijia Zhang,1 Zhengzi Yi,1 Karen L. Keung,2 Huimin Shang,3 Chengguo Wei,1 Paolo Cravedi,1 Zeguo Sun,1 Caixia Xi,1 Christopher Woytovich,1 Samira Farouk,1 Weiqing Huang,1 Khadija Banu,1 Lorenzo Gallon,4 Ciara N. Magee,5 Nader Najafian,5 Milagros Samaniego,6 Arjang Djamali ,7 Stephen I. Alexander,2 Ivy A. Rosales,8 Rex Neal Smith,8 Jenny Xiang,3 Evelyne Lerut,9 Dirk Kuypers,10,11 Maarten Naesens ,10,11 Philip J. O’Connell,2 Robert Colvin,8 Madhav C. Menon,1 and Barbara Murphy1 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background In kidney transplant recipients, surveillance biopsies can reveal, despite stable graft function, histologic features of acute rejection and borderline changes that are associated with undesirable graft outcomes. Noninvasive biomarkers of subclinical acute rejection are needed to avoid the risks and costs associated with repeated biopsies. Methods We examined subclinical histologic and functional changes in kidney transplant recipients from the prospective Genomics of Chronic Allograft Rejection (GoCAR) study who underwent surveillance biopsies over 2 years, identifying those with subclinical or borderline acute cellular rejection (ACR) at 3 months (ACR-3) post-transplant. We performed RNA sequencing on whole blood collected from 88 indi- viduals at the time of 3-month surveillance biopsy to identify transcripts associated with ACR-3, developed a novel sequencing-based targeted expression assay, and validated this gene signature in an independent cohort. -

Human Induced Pluripotent Stem Cell–Derived Podocytes Mature Into Vascularized Glomeruli Upon Experimental Transplantation
BASIC RESEARCH www.jasn.org Human Induced Pluripotent Stem Cell–Derived Podocytes Mature into Vascularized Glomeruli upon Experimental Transplantation † Sazia Sharmin,* Atsuhiro Taguchi,* Yusuke Kaku,* Yasuhiro Yoshimura,* Tomoko Ohmori,* ‡ † ‡ Tetsushi Sakuma, Masashi Mukoyama, Takashi Yamamoto, Hidetake Kurihara,§ and | Ryuichi Nishinakamura* *Department of Kidney Development, Institute of Molecular Embryology and Genetics, and †Department of Nephrology, Faculty of Life Sciences, Kumamoto University, Kumamoto, Japan; ‡Department of Mathematical and Life Sciences, Graduate School of Science, Hiroshima University, Hiroshima, Japan; §Division of Anatomy, Juntendo University School of Medicine, Tokyo, Japan; and |Japan Science and Technology Agency, CREST, Kumamoto, Japan ABSTRACT Glomerular podocytes express proteins, such as nephrin, that constitute the slit diaphragm, thereby contributing to the filtration process in the kidney. Glomerular development has been analyzed mainly in mice, whereas analysis of human kidney development has been minimal because of limited access to embryonic kidneys. We previously reported the induction of three-dimensional primordial glomeruli from human induced pluripotent stem (iPS) cells. Here, using transcription activator–like effector nuclease-mediated homologous recombination, we generated human iPS cell lines that express green fluorescent protein (GFP) in the NPHS1 locus, which encodes nephrin, and we show that GFP expression facilitated accurate visualization of nephrin-positive podocyte formation in -
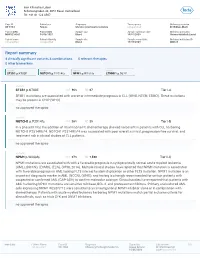
Report Summary 4 Clinically Significant Variants & Combinations 0 Relevant Therapies 0 Other Biomarkers
Inov Klinisches Labor Schützengraben 42, 4051 Basel, Switzerland Tel: +41 61 123 4567 Case ID Patient sex Diagnosis Tumor purity Ordering physician XB-1064 Female Chronic lymphocytic leukemia not specified Dr. Niklaus Marti Patient MRN Patient DOB Sample type Sample collection date Ordering institution MRN1234567 04/08/1957 Blood 10/01/2020 Universitätsklinik Liestal Patient name Patient ethnicity Sample site Sample receipt date Ordering institution ID not specified not specified Blood 10/03/2020 OID123 Report summary 4 clinically significant variants & combinations 0 relevant therapies 0 other biomarkers variant variant variant variant SF3B1 p.K700E NOTCH1 p.P2514fs NPM1 p.W288fs CTNNB1 p.D32Y variant SF3B1 p.K700E VAF 36% RD 67 Tier I-A SF3B1 mutations are associated with worse or intermediate prognosis in CLL (WHO, NCCN, ESMO). These mutations may be present in CHIP (WHO). no approved therapies variant NOTCH1 p.P2514fs VAF 28% RD 25 Tier I-B In a phase III trial, the addition of rituximab to FC chemotherapy showed no benefit in patients with CLL harboring NOTCH1 P2514Rfs*4. NOTCH1 P2514Rfs*4 was associated with poor overall survival, progression-free survival, and treatment risk in clinical studies of CLL patients. no approved therapies variant NPM1 p.W288fs VAF 27% RD 1,590 Tier II-C NPM1 mutations are associated with with a favorable prognosis in cytogenetically normal acute myeloid leukemia (AML), (NCCN), (ESMO), (ELN), (WHO, 2016). Multiple clinical studies have reported that NPM1 mutation is associated with favorable prognosis in AML lacking FLT3 internal tandem duplication or other FLT3 mutation. NPM1 mutation is an important diagnostic marker in AML (NCCN), (WHO), and testing is strongly recommended for certain patients with suspected or confirmed AML (CAP-ASH) to confirm molecular subtype. -

NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 11-7-2018 Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer Jairo Ramos [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Clinical Epidemiology Commons Recommended Citation Ramos, Jairo, "Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer" (2018). FIU Electronic Theses and Dissertations. 3872. https://digitalcommons.fiu.edu/etd/3872 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida DECIPHER MECHANISMS BY WHICH NUCLEAR RESPIRATORY FACTOR ONE (NRF1) COORDINATES CHANGES IN THE TRANSCRIPTIONAL AND CHROMATIN LANDSCAPE AFFECTING DEVELOPMENT AND PROGRESSION OF INVASIVE BREAST CANCER A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in PUBLIC HEALTH by Jairo Ramos 2018 To: Dean Tomás R. Guilarte Robert Stempel College of Public Health and Social Work This dissertation, Written by Jairo Ramos, and entitled Decipher Mechanisms by Which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer, having been approved in respect to style and intellectual content, is referred to you for judgment.