Identifying and Predicting Molecular Signatures in Glioblastoma Using Imaging-Derived
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Investigating the Genetic Basis of Cisplatin-Induced Ototoxicity in Adult South African Patients
--------------------------------------------------------------------------- Investigating the genetic basis of cisplatin-induced ototoxicity in adult South African patients --------------------------------------------------------------------------- by Timothy Francis Spracklen SPRTIM002 SUBMITTED TO THE UNIVERSITY OF CAPE TOWN In fulfilment of the requirements for the degree MSc(Med) Faculty of Health Sciences UNIVERSITY OF CAPE TOWN University18 December of Cape 2015 Town Supervisor: Prof. Rajkumar S Ramesar Co-supervisor: Ms A Alvera Vorster Division of Human Genetics, Department of Pathology, University of Cape Town 1 The copyright of this thesis vests in the author. No quotation from it or information derived from it is to be published without full acknowledgement of the source. The thesis is to be used for private study or non- commercial research purposes only. Published by the University of Cape Town (UCT) in terms of the non-exclusive license granted to UCT by the author. University of Cape Town Declaration I, Timothy Spracklen, hereby declare that the work on which this dissertation/thesis is based is my original work (except where acknowledgements indicate otherwise) and that neither the whole work nor any part of it has been, is being, or is to be submitted for another degree in this or any other university. I empower the university to reproduce for the purpose of research either the whole or any portion of the contents in any manner whatsoever. Signature: Date: 18 December 2015 ' 2 Contents Abbreviations ………………………………………………………………………………….. 1 List of figures …………………………………………………………………………………... 6 List of tables ………………………………………………………………………………….... 7 Abstract ………………………………………………………………………………………… 10 1. Introduction …………………………………………………………………………………. 11 1.1 Cancer …………………………………………………………………………….. 11 1.2 Adverse drug reactions ………………………………………………………….. 12 1.3 Cisplatin …………………………………………………………………………… 12 1.3.1 Cisplatin’s mechanism of action ……………………………………………… 13 1.3.2 Adverse reactions to cisplatin therapy ………………………………………. -

Deep Sequencing of the Human Retinae Reveals the Expression of Odorant Receptors
fncel-11-00003 January 20, 2017 Time: 14:24 # 1 CORE Metadata, citation and similar papers at core.ac.uk Provided by Frontiers - Publisher Connector ORIGINAL RESEARCH published: 24 January 2017 doi: 10.3389/fncel.2017.00003 Deep Sequencing of the Human Retinae Reveals the Expression of Odorant Receptors Nikolina Jovancevic1*, Kirsten A. Wunderlich2, Claudia Haering1, Caroline Flegel1, Désirée Maßberg1, Markus Weinrich1, Lea Weber1, Lars Tebbe2, Anselm Kampik3, Günter Gisselmann1, Uwe Wolfrum2, Hanns Hatt1† and Lian Gelis1† 1 Department of Cell Physiology, Ruhr-University Bochum, Bochum, Germany, 2 Department of Cell and Matrix Biology, Johannes Gutenberg University of Mainz, Mainz, Germany, 3 Department of Ophthalmology, Ludwig Maximilian University of Munich, Munich, Germany Several studies have demonstrated that the expression of odorant receptors (ORs) occurs in various tissues. These findings have served as a basis for functional studies that demonstrate the potential of ORs as drug targets for a clinical application. To the best of our knowledge, this report describes the first evaluation of the mRNA expression of ORs and the localization of OR proteins in the human retina that set a Edited by: stage for subsequent functional analyses. RNA-Sequencing datasets of three individual Hansen Wang, University of Toronto, Canada neural retinae were generated using Next-generation sequencing and were compared Reviewed by: to previously published but reanalyzed datasets of the peripheral and the macular Ewald Grosse-Wilde, human retina and to reference tissues. The protein localization of several ORs was Max Planck Institute for Chemical Ecology (MPG), Germany investigated by immunohistochemistry. The transcriptome analyses detected an average Takaaki Sato, of 14 OR transcripts in the neural retina, of which OR6B3 is one of the most highly National Institute of Advanced expressed ORs. -
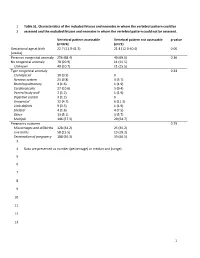
1 Table S1. Characteristics of the Included Fetuses and Neonates in Whom the Vertebral Pattern Could Be 1 Assessed and the Exclu
1 Table S1. Characteristics of the included fetuses and neonates in whom the vertebral pattern could be 2 assessed and the excluded fetuses and neonates in whom the vertebral pattern could not be assessed. Vertebral pattern assessable Vertebral pattern not assessable p-value (n=374) (n=71) Gestational age at birth 22.7 (11.9-41.3) 21.4 (12.0-40.4) 0.06 (weeks) Presence congenital anomaly 256 (68.4) 49 (69.0) 0.36 No congenital anomaly 78 (20.9) 11 (15.5) Unknown 40 (10.7) 11 (15.5) Type congenital anomaly 0.24 Craniofacial 10 (3.9) 0 Nervous system 25 (9.8) 3 (5.7) Bronchopulmonary 4 (1.6) 1 (1.9) Cardiovascular 27 (10.6) 5 (9.4) Ventral body wall 3 (1.2) 1 (1.9) Digestive system 3 (1.2) 0 Urogenital 12 (4.7) 6 (11.3) Limb defects 9 (3.5) 1 (1.9) Skeletal 4 (1.6) 4 (7.5) Other 13 (5.1) 3 (5.7) Multiple 146 (57.3) 29 (54.7) Pregnancy outcome 0.79 Miscarriages and stillbirths 128 (34.2) 25 (35.2) Live births 58 (15.5) 13 (18.3) Termination of pregnancy 188 (50.3) 33 (46.5) 3 4 Data are presented as number (percentage) or median and (range). 5 6 7 8 9 10 11 12 13 1 14 Table S2. Gestational age at birth, age at death, presence of congenital abnormalities and cause of death of the included live births. GA at birth Age at death Cervical Congenital abnormalities Cause of death (weeks) (days) rib(s) 22.3 0 No. -

Multiple Loci Modulate Opioid Therapy Response for Cancer Pain
Published OnlineFirst May 27, 2011; DOI: 10.1158/1078-0432.CCR-10-3028 Clinical Cancer Predictive Biomarkers and Personalized Medicine Research Multiple Loci Modulate Opioid Therapy Response for Cancer Pain Antonella Galvan1, Frank Skorpen2,Pal Klepstad2,3, Anne Kari Knudsen2, Torill Fladvad2, Felicia S. Falvella1, Alessandra Pigni1, Cinzia Brunelli1, Augusto Caraceni1,2, Stein Kaasa2,4, and Tommaso A. Dragani1 Abstract Purpose: Patients treated with opioid drugs for cancer pain experience different relief responses, raising the possibility that genetic factors play a role in opioid therapy outcome. In this study, we tested the hypothesis that genetic variations may control individual response to opioid drugs in cancer patients. Experimental Design: We tested 1 million single-nucleotide polymorphisms (SNP) in European cancer patients, selected in a first series, for extremely poor (pain relief 40%; n ¼ 145) or good (pain relief 90%; n ¼ 293) responses to opioid therapy using a DNA-pooling approach. Candidate SNPs identified by SNP- array were genotyped in individual samples constituting DNA pools as well as in a second series of 570 patients. Results: Association analysis in 1,008 cancer patients identified eight SNPs significantly associated with À pain relief at a statistical threshold of P < 1.0 Â 10 3, with rs12948783, upstream of the RHBDF2 gene, À showing the best statistical association (P ¼ 8.1 Â 10 9). Functional annotation analysis of SNP-tagged genes suggested the involvement of genes acting on processes of the neurologic system. Conclusion: Our results indicate that the identified SNP panel can modulate the response of cancer patients to opioid therapy and may provide a new tool for personalized therapy of cancer pain. -

Identification of Candidate Biomarkers and Pathways Associated with Type 1 Diabetes Mellitus Using Bioinformatics Analysis
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Identification of candidate biomarkers and pathways associated with type 1 diabetes mellitus using bioinformatics analysis Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.06.08.447531; this version posted June 9, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Type 1 diabetes mellitus (T1DM) is a metabolic disorder for which the underlying molecular mechanisms remain largely unclear. This investigation aimed to elucidate essential candidate genes and pathways in T1DM by integrated bioinformatics analysis. In this study, differentially expressed genes (DEGs) were analyzed using DESeq2 of R package from GSE162689 of the Gene Expression Omnibus (GEO). Gene ontology (GO) enrichment analysis, REACTOME pathway enrichment analysis, and construction and analysis of protein-protein interaction (PPI) network, modules, miRNA-hub gene regulatory network and TF-hub gene regulatory network, and validation of hub genes were then performed. A total of 952 DEGs (477 up regulated and 475 down regulated genes) were identified in T1DM. GO and REACTOME enrichment result results showed that DEGs mainly enriched in multicellular organism development, detection of stimulus, diseases of signal transduction by growth factor receptors and second messengers, and olfactory signaling pathway. -

The Potential Druggability of Chemosensory G Protein-Coupled Receptors
International Journal of Molecular Sciences Review Beyond the Flavour: The Potential Druggability of Chemosensory G Protein-Coupled Receptors Antonella Di Pizio * , Maik Behrens and Dietmar Krautwurst Leibniz-Institute for Food Systems Biology at the Technical University of Munich, Freising, 85354, Germany; [email protected] (M.B.); [email protected] (D.K.) * Correspondence: [email protected]; Tel.: +49-8161-71-2904; Fax: +49-8161-71-2970 Received: 13 February 2019; Accepted: 12 March 2019; Published: 20 March 2019 Abstract: G protein-coupled receptors (GPCRs) belong to the largest class of drug targets. Approximately half of the members of the human GPCR superfamily are chemosensory receptors, including odorant receptors (ORs), trace amine-associated receptors (TAARs), bitter taste receptors (TAS2Rs), sweet and umami taste receptors (TAS1Rs). Interestingly, these chemosensory GPCRs (csGPCRs) are expressed in several tissues of the body where they are supposed to play a role in biological functions other than chemosensation. Despite their abundance and physiological/pathological relevance, the druggability of csGPCRs has been suggested but not fully characterized. Here, we aim to explore the potential of targeting csGPCRs to treat diseases by reviewing the current knowledge of csGPCRs expressed throughout the body and by analysing the chemical space and the drug-likeness of flavour molecules. Keywords: smell; taste; flavour molecules; drugs; chemosensory receptors; ecnomotopic expression 1. Introduction Thirty-five percent of approved drugs act by modulating G protein-coupled receptors (GPCRs) [1,2]. GPCRs, also named 7-transmembrane (7TM) receptors, based on their canonical structure, are the largest family of membrane receptors in the human genome. -

Sex-Specific Transcriptome Differences in Human Adipose
G C A T T A C G G C A T genes Article Sex-Specific Transcriptome Differences in Human Adipose Mesenchymal Stem Cells 1, 2, 3 1,3 Eva Bianconi y, Raffaella Casadei y , Flavia Frabetti , Carlo Ventura , Federica Facchin 1,3,* and Silvia Canaider 1,3 1 National Laboratory of Molecular Biology and Stem Cell Bioengineering of the National Institute of Biostructures and Biosystems (NIBB)—Eldor Lab, at the Innovation Accelerator, CNR, Via Piero Gobetti 101, 40129 Bologna, Italy; [email protected] (E.B.); [email protected] (C.V.); [email protected] (S.C.) 2 Department for Life Quality Studies (QuVi), University of Bologna, Corso D’Augusto 237, 47921 Rimini, Italy; [email protected] 3 Department of Experimental, Diagnostic and Specialty Medicine (DIMES), University of Bologna, Via Massarenti 9, 40138 Bologna, Italy; fl[email protected] * Correspondence: [email protected]; Tel.: +39-051-2094114 These authors contributed equally to this work. y Received: 1 July 2020; Accepted: 6 August 2020; Published: 8 August 2020 Abstract: In humans, sexual dimorphism can manifest in many ways and it is widely studied in several knowledge fields. It is increasing the evidence that also cells differ according to sex, a correlation still little studied and poorly considered when cells are used in scientific research. Specifically, our interest is on the sex-related dimorphism on the human mesenchymal stem cells (hMSCs) transcriptome. A systematic meta-analysis of hMSC microarrays was performed by using the Transcriptome Mapper (TRAM) software. This bioinformatic tool was used to integrate and normalize datasets from multiple sources and allowed us to highlight chromosomal segments and genes differently expressed in hMSCs derived from adipose tissue (hADSCs) of male and female donors. -

WO 2019/068007 Al Figure 2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/068007 Al 04 April 2019 (04.04.2019) W 1P O PCT (51) International Patent Classification: (72) Inventors; and C12N 15/10 (2006.01) C07K 16/28 (2006.01) (71) Applicants: GROSS, Gideon [EVIL]; IE-1-5 Address C12N 5/10 (2006.0 1) C12Q 1/6809 (20 18.0 1) M.P. Korazim, 1292200 Moshav Almagor (IL). GIBSON, C07K 14/705 (2006.01) A61P 35/00 (2006.01) Will [US/US]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., C07K 14/725 (2006.01) P.O. Box 4044, 7403635 Ness Ziona (TL). DAHARY, Dvir [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (21) International Application Number: Box 4044, 7403635 Ness Ziona (IL). BEIMAN, Merav PCT/US2018/053583 [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (22) International Filing Date: Box 4044, 7403635 Ness Ziona (E.). 28 September 2018 (28.09.2018) (74) Agent: MACDOUGALL, Christina, A. et al; Morgan, (25) Filing Language: English Lewis & Bockius LLP, One Market, Spear Tower, SanFran- cisco, CA 94105 (US). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/564,454 28 September 2017 (28.09.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/649,429 28 March 2018 (28.03.2018) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (71) Applicant: IMMP ACT-BIO LTD. -

Sean Raspet – Molecules
1. Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Molecular weight: 240.39 g/mol Volume (cubic Angstroems): 258.88 Atoms number (non-hydrogen): 17 miLogP: 4.43 Structure: Biological Properties: Predicted Druglikenessi: GPCR ligand -0.23 Ion channel modulator -0.03 Kinase inhibitor -0.6 Nuclear receptor ligand 0.15 Protease inhibitor -0.28 Enzyme inhibitor 0.15 Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Predicted Olfactory Receptor Activityii: OR2L13 83.715% OR1G1 82.761% OR10J5 80.569% OR2W1 78.180% OR7A2 77.696% 2. Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Molecular weight: 239.36 Volume (cubic Angstroems): 252.83 Atoms number (non-hydrogen): 17 miLogP: 4.33 Structure: Biological Properties: Predicted Druglikeness: GPCR ligand -0.6 Ion channel modulator -0.41 Kinase inhibitor -0.93 Nuclear receptor ligand -0.17 Protease inhibitor -0.39 Enzyme inhibitor 0.01 Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Predicted Olfactory Receptor Activity: OR52D1 71.900% OR1G1 70.394% 0R52I2 70.392% OR52I1 70.390% OR2Y1 70.378% 3. Commercial name: Hyperflor© IUPAC Name: 2-benzyl-1,3-dioxan-5-one SMILES: O=C1COC(CC2=CC=CC=C2)OC1 Molecular weight: 192.21 g/mol Volume -

Genetic Characterization of Greek Population Isolates Reveals Strong Genetic Drift at Missense and Trait-Associated Variants
ARTICLE Received 22 Apr 2014 | Accepted 22 Sep 2014 | Published 6 Nov 2014 DOI: 10.1038/ncomms6345 OPEN Genetic characterization of Greek population isolates reveals strong genetic drift at missense and trait-associated variants Kalliope Panoutsopoulou1,*, Konstantinos Hatzikotoulas1,*, Dionysia Kiara Xifara2,3, Vincenza Colonna4, Aliki-Eleni Farmaki5, Graham R.S. Ritchie1,6, Lorraine Southam1,2, Arthur Gilly1, Ioanna Tachmazidou1, Segun Fatumo1,7,8, Angela Matchan1, Nigel W. Rayner1,2,9, Ioanna Ntalla5,10, Massimo Mezzavilla1,11, Yuan Chen1, Chrysoula Kiagiadaki12, Eleni Zengini13,14, Vasiliki Mamakou13,15, Antonis Athanasiadis16, Margarita Giannakopoulou17, Vassiliki-Eirini Kariakli5, Rebecca N. Nsubuga18, Alex Karabarinde18, Manjinder Sandhu1,8, Gil McVean2, Chris Tyler-Smith1, Emmanouil Tsafantakis12, Maria Karaleftheri16, Yali Xue1, George Dedoussis5 & Eleftheria Zeggini1 Isolated populations are emerging as a powerful study design in the search for low-frequency and rare variant associations with complex phenotypes. Here we genotype 2,296 samples from two isolated Greek populations, the Pomak villages (HELIC-Pomak) in the North of Greece and the Mylopotamos villages (HELIC-MANOLIS) in Crete. We compare their genomic characteristics to the general Greek population and establish them as genetic isolates. In the MANOLIS cohort, we observe an enrichment of missense variants among the variants that have drifted up in frequency by more than fivefold. In the Pomak cohort, we find novel associations at variants on chr11p15.4 showing large allele frequency increases (from 0.2% in the general Greek population to 4.6% in the isolate) with haematological traits, for example, with mean corpuscular volume (rs7116019, P ¼ 2.3 Â 10 À 26). We replicate this association in a second set of Pomak samples (combined P ¼ 2.0 Â 10 À 36). -

Functional and Structural Characterization of Olfactory Receptors in Human Heart and Eye
DISSERTATION to obtain the degree Doctor Rerum Naturalium (Dr.rer.nat.) at the Faculty of Biology and Biotechnology International Graduate School Biosciences Ruhr-University Bochum Functional and structural characterization of olfactory receptors in human heart and eye Department of Cellphysiology submitted by Nikolina Jovancevic from Zadar, Croatia Bochum February 2016 First Referee: Prof. Dr. Dr. Dr. Hanns Hatt Second Referee: Prof. Dr. Stefan Wiese DISSERTATION zur Erlangung des Grades eines Doktors der Naturwissenschaften der Fakultät für Biologie und Biotechnologie an der Internationalen Graduiertenschule Biowissenschaften der Ruhr-Universität Bochum Funktionale und strukturelle Charakterisierung olfaktorischer Rezeptoren im humanen Herzen und Auge Lehrstuhl für Zellphysiologie vorgelegt von Nikolina Jovancevic aus Zadar, Kroatien Bochum Februar 2016 Referent: Prof. Dr. Dr. Dr. Hanns Hatt Korreferent: Prof. Dr. Stefan Wiese To my family TABLE OF CONTENTS TABEL OF CONTENT 1 INTRODUCTION 1 1.1 G protein-coupled receptors 1 1.1.1 General 1 1.1.2 Structure and classification 2 1.1.3 Olfactory Receptors 4 1.2 Function of olfactory receptors 9 1.2.1 The olfactory system 9 1.2.2 Ectopic expression of olfactory receptors 11 1.3 Excursus: Anatomy and physiology of the heart 13 1.3.1 Anatomy of the heart and blood circuit 14 1.3.2 The cardiac conduction system 15 1.3.3 Excitation-contraction coupling 16 1.3.4 Cardiac GPCRs: Modulation of cardiac contraction 17 1.4 Excursus: Anatomy and physiology of the eye 18 1.4.1 Anatomy of the retina 19