Genetic Heterogeneity in Autism Spectrum Disorders in a Population Isolate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Federal Hansard Acronyms List Remember: Ctrl+F for Quick Searches
Federal Hansard Acronyms List Remember: Ctrl+F for quick searches A B C D E F G H I J K L M N O P Q R S T U V W X Y Z A 2.5G [the first packet overlays on 2G networks] 2G second generation [the first generation of digital cellular networks, as opposed to analog] 3G third generation [next generation of cellular networks] 3GPP 3G Partnership Project [global standards body to oversee 3G] 4D meat from dead, dying, diseased or disabled animals 4GL fourth-generation language [computers] A&C automation and control A&D admission and disposition; alcohol and drugs A&E accident and emergency A&RMC formerly Austin & Repatriation Medical Centre [now Austin Health] AA anti-aircraft; Alcoholics Anonymous; Athletics Australia AAA Agriculture Advancing Australia; Australian Automobile Association; Australian Archaeological Association; Australian Airports Association AAAA Aerial Agricultural Association of Australia AAAE Australian Association of Automotive Electricians AAAGP Australian Association of Academic General Practice AAALAC Association for the Assessment and Accreditation of Laboratory Animal Care International AAB Australian Associated Brewers AAC Aboriginal advisory committee; Australian Arabic Council; AARNet Advisory Committee AACAP ATSIC-Army Community Assistance Program AACC Aboriginal Affairs Coordinating Committee [WA]; Australian Association of Career Counsellors AACM Australian Association for Computational Mechanics AACS Australian Associations of Christian Schools [note: Associations—plural] AACV Australian Association of Cattle Veterinarians AAD Australian Antarctic Division [Department of the Environment and Heritage] AADCP ASEAN-Australia Development Cooperation Program [taking over AAECP] AADS advanced air defence simulator AADT average annual daily traffic AaE Australian air Express Pty Ltd AAEC Antarctic Animal Ethics Committee AAECP ASEAN-Australia Economic Cooperation Program [finishes in 2005] AAFCANS Army and Air Force Canteen Service [now known as Frontline Defence Services] AAGP Australian Association of Group Psychotherapists Inc. -

Latent Class Analysis to Classify Patients with Transthyretin Amyloidosis by Signs and Symptoms
Neurol Ther DOI 10.1007/s40120-015-0028-y ORIGINAL RESEARCH Latent Class Analysis to Classify Patients with Transthyretin Amyloidosis by Signs and Symptoms Jose Alvir . Michelle Stewart . Isabel Conceic¸a˜o To view enhanced content go to www.neurologytherapy-open.com Received: February 21, 2015 Ó The Author(s) 2015. This article is published with open access at Springerlink.com ABSTRACT status measures for the latent classes were examined. Introduction: The aim of this study was to Results: A four-class latent class solution was develop an empirical approach to classifying thebestfitforthedata.Thelatentclasseswere patients with transthyretin amyloidosis (ATTR) characterized by the predominant symptoms based on clinical signs and symptoms. as severe neuropathy/severe autonomic, Methods: Data from 971 symptomatic subjects moderate to severe neuropathy/low to enrolled in the Transthyretin Amyloidosis moderate autonomic involvement, severe Outcomes Survey were analyzed using a latent cardiac, and moderate to severe neuropathy. class analysis approach. Differences in health Incorporating disease duration improved the model fit. It was found that measures of health Electronic supplementary material The online status varied by latent class in interpretable version of this article (doi:10.1007/s40120-015-0028-y) contains supplementary material, which is available to patterns. authorized users. Conclusion: This latent class analysis approach On behalf of the THAOS Investigators. offered promise in categorizing patients with ATTR across the spectrum of disease. The four- Trial registration: ClinicalTrials.gov number, class latent class solution included disease NCT00628745. duration and enabled better detection of J. Alvir (&) heterogeneity within and across genotypes Statistical Center for Outcomes, Real-World and than previous approaches, which have tended Aggregate Data, Global Medicines Development, Global Innovative Pharma Business, Pfizer, Inc., to classify patients a priori into neuropathic, New York, NY, USA cardiac, and mixed groups. -

Annona Coriacea Mart. Fractions Promote Cell Cycle Arrest and Inhibit Autophagic Flux in Human Cervical Cancer Cell Lines
molecules Article Annona coriacea Mart. Fractions Promote Cell Cycle Arrest and Inhibit Autophagic Flux in Human Cervical Cancer Cell Lines Izabela N. Faria Gomes 1,2, Renato J. Silva-Oliveira 2 , Viviane A. Oliveira Silva 2 , Marcela N. Rosa 2, Patrik S. Vital 1 , Maria Cristina S. Barbosa 3,Fábio Vieira dos Santos 3 , João Gabriel M. Junqueira 4, Vanessa G. P. Severino 4 , Bruno G Oliveira 5, Wanderson Romão 5, Rui Manuel Reis 2,6,7,* and Rosy Iara Maciel de Azambuja Ribeiro 1,* 1 Experimental Pathology Laboratory, Federal University of São João del Rei—CCO/UFSJ, Divinópolis 35501-296, Brazil; [email protected] (I.N.F.G.); [email protected] (P.S.V.) 2 Molecular Oncology Research Center, Barretos Cancer Hospital, Barretos 14784-400, Brazil; [email protected] (R.J.S.-O.); [email protected] (V.A.O.S.); [email protected] (M.N.R.) 3 Laboratory of Cell Biology and Mutagenesis, Federal University of São João del Rei—CCO/UFSJ, Divinópolis 35501-296, Brazil; [email protected] (M.C.S.B.); [email protected] (F.V.d.S.) 4 Special Academic Unit of Physics and Chemistry, Federal University of Goiás, Catalão 75704-020, Brazil; [email protected] (J.G.M.J.); [email protected] (V.G.P.S.) 5 Petroleomic and forensic chemistry laboratory, Department of Chemistry, Federal Institute of Espirito Santo, Vitória, ES 29075-910, Brazil; [email protected] (B.G.O.); [email protected] (W.R.) 6 Life and Health Sciences Research Institute (ICVS), Medical School, University of Minho, 4710-057 Braga, Portugal 7 3ICVS/3B’s-PT Government Associate Laboratory, 4710-057 Braga, Portugal * Correspondence: [email protected] (R.M.R.); [email protected] (R.I.M.d.A.R.); Tel.: +55-173-321-6600 (R.M.R.); +55-3736-904-484 or +55-3799-1619-155 (R.I.M.d.A.R.) Received: 25 September 2019; Accepted: 29 October 2019; Published: 1 November 2019 Abstract: Plant-based compounds are an option to explore and perhaps overcome the limitations of current antitumor treatments. -

DNV Classification Note 31.2 Strength Analysis of Hull Structure in Roll
CLASSIFICATION NOTES No. 31.2 STRENGTH ANALYSIS OF HULL STRUCTURE IN ROLL ON/ROLL OFF SHIPS AND CAR CARRIERS APRIL 2011 The content of this service document is the subject of intellectual property rights reserved by Det Norske Veritas AS (DNV). The user accepts that it is prohibited by anyone else but DNV and/or its licensees to offer and/or perform classification, certification and/or verification services, including the issuance of certificates and/or declarations of conformity, wholly or partly, on the basis of and/or pursuant to this document whether free of charge or chargeable, without DNV's prior written consent. DNV is not responsible for the consequences arising from any use of this document by others. DET NORSKE VERITAS Veritasveien 1, NO-1322 Høvik, Norway Tel.: +47 67 57 99 00 Fax: +47 67 57 99 11 FOREWORD DET NORSKE VERITAS (DNV) is an autonomous and independent foundation with the objectives of safeguarding life, property and the environment, at sea and onshore. DNV undertakes classification, certification, and other verification and consultancy services relating to quality of ships, offshore units and installations, and onshore industries worldwide, and carries out research in relation to these functions. Classification Notes Classification Notes are publications that give practical information on classification of ships and other objects. Examples of design solutions, calculation methods, specifications of test procedures, as well as acceptable repair methods for some components are given as interpretations of the more general rule requirements. All publications may be downloaded from the Society’s Web site http://www.dnv.com/. The Society reserves the exclusive right to interpret, decide equivalence or make exemptions to this Classification Note. -

A Chromosome Level Genome of Astyanax Mexicanus Surface Fish for Comparing Population
bioRxiv preprint doi: https://doi.org/10.1101/2020.07.06.189654; this version posted July 6, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Title 2 A chromosome level genome of Astyanax mexicanus surface fish for comparing population- 3 specific genetic differences contributing to trait evolution. 4 5 Authors 6 Wesley C. Warren1, Tyler E. Boggs2, Richard Borowsky3, Brian M. Carlson4, Estephany 7 Ferrufino5, Joshua B. Gross2, LaDeana Hillier6, Zhilian Hu7, Alex C. Keene8, Alexander Kenzior9, 8 Johanna E. Kowalko5, Chad Tomlinson10, Milinn Kremitzki10, Madeleine E. Lemieux11, Tina 9 Graves-Lindsay10, Suzanne E. McGaugh12, Jeff T. Miller12, Mathilda Mommersteeg7, Rachel L. 10 Moran12, Robert Peuß9, Edward Rice1, Misty R. Riddle13, Itzel Sifuentes-Romero5, Bethany A. 11 Stanhope5,8, Clifford J. Tabin13, Sunishka Thakur5, Yamamoto Yoshiyuki14, Nicolas Rohner9,15 12 13 Authors for correspondence: Wesley C. Warren ([email protected]), Nicolas Rohner 14 ([email protected]) 15 16 Affiliation 17 1Department of Animal Sciences, Department of Surgery, Institute for Data Science and 18 Informatics, University of Missouri, Bond Life Sciences Center, Columbia, MO 19 2 Department of Biological Sciences, University of Cincinnati, Cincinnati, OH 20 3 Department of Biology, New York University, New York, NY 21 4 Department of Biology, The College of Wooster, Wooster, OH 22 5 Harriet L. Wilkes Honors College, Florida Atlantic University, Jupiter FL 23 6 Department of Genome Sciences, University of Washington, Seattle, WA 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.07.06.189654; this version posted July 6, 2020. -

Sphingomyelin Synthases in Neuropsychiatric Health and Disease
Neurosignals 2019;27(S1):54-76 DOI: 10.33594/00000020010.33594/000000200 © 2019 The Author(s).© 2019 Published The Author(s) by Published online: 27 DecemberDecember 20192019 Cell Physiol BiochemPublished Press GmbH&Co. by Cell Physiol KG Biochem 54 Press GmbH&Co. KG, Duesseldorf Mühle et al.: SMS in Neuropsychiatry Accepted: 23 December 2019 www.neuro-signals.com This article is licensed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 Interna- tional License (CC BY-NC-ND). Usage and distribution for commercial purposes as well as any distribution of modified material requires written permission. Review Sphingomyelin Synthases in Neuropsychiatric Health and Disease Christiane Mühle Roberto D. Bilbao Canalejas Johannes Kornhuber Department of Psychiatry and Psychotherapy, Friedrich-Alexander University Erlangen-Nürnberg (FAU), Erlangen, Germany Key Words Sphingomyelin synthase • Neurological disease • Psychiatric disease • Brain • Central nervous system Abstract Sphingomyelin synthases (SMS) catalyze the conversion of ceramide and phosphatidylcholine to sphingomyelin and diacylglycerol and are thus crucial for the balance between synthesis and degradation of these structural and bioactive molecules. SMS thereby play an essential role in sphingolipid metabolism, cell signaling, proliferation and differentiation processes. Although tremendous progress has been made toward understanding the involvement of SMS in physiological and pathological processes, literature in the area of neuropsychiatry is still limited. In this review, we summarize the main features of SMS as well as the current methodologies and tools used for their study and provide an overview of SMS in the central nervous system and their implications in neurological as well as psychiatric disorders. This way, we aim at establishing a basis for future mechanistic as well as clinical investigations on SMS in neuropsychiatric health and diseases. -

Integrative Genomic Characterization Identifies Molecular Subtypes of Lung Carcinoids
Published OnlineFirst July 12, 2019; DOI: 10.1158/0008-5472.CAN-19-0214 Cancer Genome and Epigenome Research Integrative Genomic Characterization Identifies Molecular Subtypes of Lung Carcinoids Saurabh V. Laddha1, Edaise M. da Silva2, Kenneth Robzyk2, Brian R. Untch3, Hua Ke1, Natasha Rekhtman2, John T. Poirier4, William D. Travis2, Laura H. Tang2, and Chang S. Chan1,5 Abstract Lung carcinoids (LC) are rare and slow growing primary predominately found at peripheral and endobronchial lung, lung neuroendocrine tumors. We performed targeted exome respectively. The LC3 subtype was diagnosed at a younger age sequencing, mRNA sequencing, and DNA methylation array than LC1 and LC2 subtypes. IHC staining of two biomarkers, analysis on macro-dissected LCs. Recurrent mutations were ASCL1 and S100, sufficiently stratified the three subtypes. enriched for genes involved in covalent histone modification/ This molecular classification of LCs into three subtypes may chromatin remodeling (34.5%; MEN1, ARID1A, KMT2C, and facilitate understanding of their molecular mechanisms and KMT2A) as well as DNA repair (17.2%) pathways. Unsuper- improve diagnosis and clinical management. vised clustering and principle component analysis on gene expression and DNA methylation profiles showed three robust Significance: Integrative genomic analysis of lung carcinoids molecular subtypes (LC1, LC2, LC3) with distinct clinical identifies three novel molecular subtypes with distinct clinical features. MEN1 gene mutations were found to be exclusively features and provides insight into their distinctive molecular enriched in the LC2 subtype. LC1 and LC3 subtypes were signatures of tumorigenesis, diagnosis, and prognosis. Introduction of Ki67 between ACs and TCs does not enable reliable stratification between well-differentiated LCs (6, 7). -
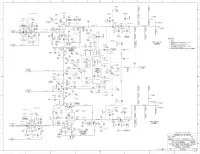
ML19092A066.Pdf
r .. - .. - .. - .. - .. - .. - .. 8.- .. -.. -.. -.. -.. -.. -r- .. -.. -.. -.. -.. -.. -i .. -.. -.. -.. -.. -.. -.. I.. -.. -.. -.. -.. -.. -.. 6.- .. -.. -.. -.. -.. -.. -1- .. -.. -.. -.. -.. -.. -s- .. -.. -.. -.. -.. -.. -. f .- .. -.. -.. -.. -.. -.. 4.- .. -.. -.. -.. -.. -.. -1- .. -.. -.. -.. -.. -.. -3- .. - .. - .. - .. - .. - .. - .. I.. -.. -.. -.. -.. -.. -.. 2.- .. -.. -.. -.. -.. -.. -1- .. -.. -.. -.. -.. -.. -·1- .. -.. - .. - .. - .. - .. - .. l CSAS ____ I 5OPENS ' ' ' [] ~® H v --017 ' I Cl S-A CLOSES TEST ;--- -s ~---<0 CONN. & DRAIN CSAS I ~---: ' STARTS I n ' I' ' I r- * 0 ' ' u 048-HCB-4 ' ' I C I NOZZLE! ' ITJ : i ~....L..._t-~::---1~------ -(0 TEST ' ' 'i' CONN. / NOTES 2 & 3 l..'..J~ ' A 0 ' u n r;ID ---~---:-n__ I PENI2Jl A I CONTAINMENT HV L 1 '---"'017 SPFiiAY PUMP (FE "' ~ -·-· v ~ 017 ,'Y :': r: . 0006 .. 7_ I' I _, 1- \@005 ' ' M-! 2GL01 ~- --- J-, ~ .. !i! L--,,.-,-_l r- --- I SEE < GCB HCB I ~ 049-HCB-2 l/2 I I NOTE 5 ( Vl ( c- 7J TO OA0009 1/ C I NOZZLE! ~---~ : "'"8 n ( B- 61 -(0 ?-:-- I P-eg I ::.. r04!-HCB-!0 - ~ I '-I L_~-L.....J~==---1CJf;J------ ~ B'X 10' lr-----~0~03~-~G~C=B--1~0~------------------r-----~,-~~~~~~0~40~-~H=C=B~-1,0~--..--~~,~--L~~,-~~D<}--------.------L-. __ ~r- 041-HCB-6 r.__ I /2' T. C. [- ~ :, I > ~002) f::J- v:~~~4 II LJ 0:0--:-HCB-3/~ • V0~ V00~4 "'iii .__ ~ 1\ ~ ~ ... 1,'! l~]' \_ 041 -HCB-2 I /2 3/8' . j ... ,,.. ~_j l' X 314'- ._- 040-HCB-1 .;,;;j I 3 NOZZLES>' ~ t ~ ~ ss \ ,7 i!l 1 s \'"' •I" ~ Vlilt 28 - -~ Ur-...~ ~: TUB! NG- --._ ~ ..-,~I"~ _'I"~ ~ Io G 2 001- HCB-/14 0 GCB GCD ;7 ~ ;7 M • •z CTMT. RECI RC. 0 ~R\~~ 8 u .... _ I S 00 u SUMP 7~ C) 1"(0) ., "':I: <S>N u 3/ 4' " ., ~ hrl!:'l .._ ~ I 2' X I 4' ~tA' ~> • M-1 2EJ"I L,'> ~ FILL VENT CD > II <f 0 M ~;~~ u c E- 8> V0002 NOTE 4 --;= I . -

Immune Suppressive Landscape in the Human Esophageal Squamous Cell
ARTICLE https://doi.org/10.1038/s41467-020-20019-0 OPEN Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment ✉ Yingxia Zheng 1,2,10 , Zheyi Chen1,10, Yichao Han 3,10, Li Han1,10, Xin Zou4, Bingqian Zhou1, Rui Hu5, Jie Hao4, Shihao Bai4, Haibo Xiao5, Wei Vivian Li6, Alex Bueker7, Yanhui Ma1, Guohua Xie1, Junyao Yang1, ✉ ✉ ✉ Shiyu Chen1, Hecheng Li 3 , Jian Cao 7,8 & Lisong Shen 1,9 1234567890():,; Cancer immunotherapy has revolutionized cancer treatment, and it relies heavily on the comprehensive understanding of the immune landscape of the tumor microenvironment (TME). Here, we obtain a detailed immune cell atlas of esophageal squamous cell carcinoma (ESCC) at single-cell resolution. Exhausted T and NK cells, regulatory T cells (Tregs), alternatively activated macrophages and tolerogenic dendritic cells are dominant in the TME. Transcriptional profiling coupled with T cell receptor (TCR) sequencing reveal lineage con- nections in T cell populations. CD8 T cells show continuous progression from pre-exhausted to exhausted T cells. While exhausted CD4, CD8 T and NK cells are major proliferative cell components in the TME, the crosstalk between macrophages and Tregs contributes to potential immunosuppression in the TME. Our results indicate several immunosuppressive mechanisms that may be simultaneously responsible for the failure of immuno-surveillance. Specific targeting of these immunosuppressive pathways may reactivate anti-tumor immune responses in ESCC. 1 Department of Laboratory Medicine, Xin Hua Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China. 2 Institute of Biliary Tract Diseases Research, Shanghai Jiao Tong University School of Medicine, Shanghai, China. 3 Department of Thoracic Surgery, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China. -

LUCAS 2018 Technical Reference Document C3 Classification (Land
Regional statistics and Geographic Information Author: E4.LUCAS (ESTAT) TechnicalDocuments 2018 LUCAS 2018 (Land Use / Cover Area Frame Survey) Technical reference document C3 Classification (Land cover & Land use) Regional statistics and Geographic Information Author: E4.LUCAS (ESTAT) TechnicalDocuments 2018 Table of Contents 1 Scope and Introduction ............................................................................................................................. 6 LUCAS survey classification comparison 2009 - 2012 ................................................................................... 7 LUCAS survey classification comparison 2012 - 2015 ................................................................................... 7 LUCAS survey classification comparison 2015 – 2018 ................................................................................... 9 Land cover and land use: general explications .............................................................................................. 9 Specific to the LUCAS classification ............................................................................................................. 10 The basic unit and the extended window of observation ........................................................................... 10 2 Land Cover Classification (LUCAS SU LC) ................................................................................................. 11 A00 ARTIFICIAL LAND ............................................................................................................................. -

Downloaded Per Proteome Cohort Via the Web- Site Links of Table 1, Also Providing Information on the Deposited Spectral Datasets
www.nature.com/scientificreports OPEN Assessment of a complete and classifed platelet proteome from genome‑wide transcripts of human platelets and megakaryocytes covering platelet functions Jingnan Huang1,2*, Frauke Swieringa1,2,9, Fiorella A. Solari2,9, Isabella Provenzale1, Luigi Grassi3, Ilaria De Simone1, Constance C. F. M. J. Baaten1,4, Rachel Cavill5, Albert Sickmann2,6,7,9, Mattia Frontini3,8,9 & Johan W. M. Heemskerk1,9* Novel platelet and megakaryocyte transcriptome analysis allows prediction of the full or theoretical proteome of a representative human platelet. Here, we integrated the established platelet proteomes from six cohorts of healthy subjects, encompassing 5.2 k proteins, with two novel genome‑wide transcriptomes (57.8 k mRNAs). For 14.8 k protein‑coding transcripts, we assigned the proteins to 21 UniProt‑based classes, based on their preferential intracellular localization and presumed function. This classifed transcriptome‑proteome profle of platelets revealed: (i) Absence of 37.2 k genome‑ wide transcripts. (ii) High quantitative similarity of platelet and megakaryocyte transcriptomes (R = 0.75) for 14.8 k protein‑coding genes, but not for 3.8 k RNA genes or 1.9 k pseudogenes (R = 0.43–0.54), suggesting redistribution of mRNAs upon platelet shedding from megakaryocytes. (iii) Copy numbers of 3.5 k proteins that were restricted in size by the corresponding transcript levels (iv) Near complete coverage of identifed proteins in the relevant transcriptome (log2fpkm > 0.20) except for plasma‑derived secretory proteins, pointing to adhesion and uptake of such proteins. (v) Underrepresentation in the identifed proteome of nuclear‑related, membrane and signaling proteins, as well proteins with low‑level transcripts. -

Manuel Du HCB Pour L'utilisation Confinée D'organismes
Manuel du HCB pour l’utilisation confinée d’organismes génétiquement modifiés Rueda CC IRRI CC IRRI Mafunyane CC Mafunyane Haut Conseil des biotechnologies 244 boulevard Saint-Germain 75007 Paris Remerciements Ce document est le résultat d'un travail collectif des membres du collège confiné1 du Comité scientifique du Haut Conseil des biotechnologies (HCB) lors de son premier mandat (2009- 2014) 2 , composé de : Jean-Christophe Pagès, Président, Jean-Jacques Leguay, Vice-Président, Elie Dassa, coordinateur, et par ordre alphabétique : Claude Bagnis, Pascal Boireau, Jean-Luc Darlix, Hubert de Verneuil, Robert Drillien, Anne Dubart-Kupperschmitt, Claudine Franche, Philippe Guerche, André Jestin, Bernard Klonjkowski, Olivier Le Gall, Didier Lereclus, Daniel Parzy, Patrick Saindrenan, Pascal Simonet et Jean-Luc Vilotte. Le Comité scientifique du HCB tient à remercier pour leur relecture critique, l'association Organibio, M. Bernard Cornillon (INSERM, risques biologiques). Le HCB remercie par ailleurs les experts qui ont contribué à la rédaction de l'ouvrage "PRINCIPES DE CLASSEMENT ET GUIDES OFFICIELS DE LA COMMISSION DE GENIE GENETIQUE" (publié en 1993 sous le double timbre du Ministère de la Recherche et du Ministère de l’Aménagement du Territoire et de l’Environnement), document dont la teneur a largement inspiré la rédaction du présent manuel. 1 Sous-ensemble d’experts du Comité scientifique traitant des questions spécifiques aux biotechnologies destinées à un usage en milieu confiné. 2 L’équipe du HCB tient à remercier Marion Pillot (Chargée de mission) pour son travail éditorial sur la première version du manuel. Avant-propos du Président du Comité scientifique du HCB Le présent manuel, supervisé par le CS du HCB, est la deuxième version d’un texte d’aide à la déclaration de l’usage d’OGM dans un environnement confiné.