Usda Bioinformatics Server Training Usda Bioinformatics Server Training
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Fundamentals of UNIX Lab 5.4.6 – Listing Directory Information (Estimated Time: 30 Min.)
Fundamentals of UNIX Lab 5.4.6 – Listing Directory Information (Estimated time: 30 min.) Objectives: • Learn to display directory and file information • Use the ls (list files) command with various options • Display hidden files • Display files and file types • Examine and interpret the results of a long file listing • List individual directories • List directories recursively Background: In this lab, the student will use the ls command, which is used to display the contents of a directory. This command will display a listing of all files and directories within the current directory or specified directory or directories. If no pathname is given as an argument, ls will display the contents of the current directory. The ls command will list any subdirectories and files that are in the current working directory if a pathname is specified. The ls command will also default to a wide listing and display only file and directory names. There are many options that can be used with the ls command, which makes this command one of the more flexible and useful UNIX commands. Command Format: ls [-option(s)] [pathname[s]] Tools / Preparation: a) Before starting this lab, the student should review Chapter 5, Section 4 – Listing Directory Contents b) The student will need the following: 1. A login user ID, for example user2, and a password assigned by their instructor. 2. A computer running the UNIX operating system with CDE. 3. Networked computers in classroom. Notes: 1 - 5 Fundamentals UNIX 2.0—-Lab 5.4.6 Copyright 2002, Cisco Systems, Inc. Use the diagram of the sample Class File system directory tree to assist with this lab. -

Copy on Write Based File Systems Performance Analysis and Implementation
Copy On Write Based File Systems Performance Analysis And Implementation Sakis Kasampalis Kongens Lyngby 2010 IMM-MSC-2010-63 Technical University of Denmark Department Of Informatics Building 321, DK-2800 Kongens Lyngby, Denmark Phone +45 45253351, Fax +45 45882673 [email protected] www.imm.dtu.dk Abstract In this work I am focusing on Copy On Write based file systems. Copy On Write is used on modern file systems for providing (1) metadata and data consistency using transactional semantics, (2) cheap and instant backups using snapshots and clones. This thesis is divided into two main parts. The first part focuses on the design and performance of Copy On Write based file systems. Recent efforts aiming at creating a Copy On Write based file system are ZFS, Btrfs, ext3cow, Hammer, and LLFS. My work focuses only on ZFS and Btrfs, since they support the most advanced features. The main goals of ZFS and Btrfs are to offer a scalable, fault tolerant, and easy to administrate file system. I evaluate the performance and scalability of ZFS and Btrfs. The evaluation includes studying their design and testing their performance and scalability against a set of recommended file system benchmarks. Most computers are already based on multi-core and multiple processor architec- tures. Because of that, the need for using concurrent programming models has increased. Transactions can be very helpful for supporting concurrent program- ming models, which ensure that system updates are consistent. Unfortunately, the majority of operating systems and file systems either do not support trans- actions at all, or they simply do not expose them to the users. -
Administering Unidata on UNIX Platforms
C:\Program Files\Adobe\FrameMaker8\UniData 7.2\7.2rebranded\ADMINUNIX\ADMINUNIXTITLE.fm March 5, 2010 1:34 pm Beta Beta Beta Beta Beta Beta Beta Beta Beta Beta Beta Beta Beta Beta Beta Beta UniData Administering UniData on UNIX Platforms UDT-720-ADMU-1 C:\Program Files\Adobe\FrameMaker8\UniData 7.2\7.2rebranded\ADMINUNIX\ADMINUNIXTITLE.fm March 5, 2010 1:34 pm Beta Beta Beta Beta Beta Beta Beta Beta Beta Beta Beta Beta Beta Notices Edition Publication date: July, 2008 Book number: UDT-720-ADMU-1 Product version: UniData 7.2 Copyright © Rocket Software, Inc. 1988-2010. All Rights Reserved. Trademarks The following trademarks appear in this publication: Trademark Trademark Owner Rocket Software™ Rocket Software, Inc. Dynamic Connect® Rocket Software, Inc. RedBack® Rocket Software, Inc. SystemBuilder™ Rocket Software, Inc. UniData® Rocket Software, Inc. UniVerse™ Rocket Software, Inc. U2™ Rocket Software, Inc. U2.NET™ Rocket Software, Inc. U2 Web Development Environment™ Rocket Software, Inc. wIntegrate® Rocket Software, Inc. Microsoft® .NET Microsoft Corporation Microsoft® Office Excel®, Outlook®, Word Microsoft Corporation Windows® Microsoft Corporation Windows® 7 Microsoft Corporation Windows Vista® Microsoft Corporation Java™ and all Java-based trademarks and logos Sun Microsystems, Inc. UNIX® X/Open Company Limited ii SB/XA Getting Started The above trademarks are property of the specified companies in the United States, other countries, or both. All other products or services mentioned in this document may be covered by the trademarks, service marks, or product names as designated by the companies who own or market them. License agreement This software and the associated documentation are proprietary and confidential to Rocket Software, Inc., are furnished under license, and may be used and copied only in accordance with the terms of such license and with the inclusion of the copyright notice. -

System Calls System Calls
System calls We will investigate several issues related to system calls. Read chapter 12 of the book Linux system call categories file management process management error handling note that these categories are loosely defined and much is behind included, e.g. communication. Why? 1 System calls File management system call hierarchy you may not see some topics as part of “file management”, e.g., sockets 2 System calls Process management system call hierarchy 3 System calls Error handling hierarchy 4 Error Handling Anything can fail! System calls are no exception Try to read a file that does not exist! Error number: errno every process contains a global variable errno errno is set to 0 when process is created when error occurs errno is set to a specific code associated with the error cause trying to open file that does not exist sets errno to 2 5 Error Handling error constants are defined in errno.h here are the first few of errno.h on OS X 10.6.4 #define EPERM 1 /* Operation not permitted */ #define ENOENT 2 /* No such file or directory */ #define ESRCH 3 /* No such process */ #define EINTR 4 /* Interrupted system call */ #define EIO 5 /* Input/output error */ #define ENXIO 6 /* Device not configured */ #define E2BIG 7 /* Argument list too long */ #define ENOEXEC 8 /* Exec format error */ #define EBADF 9 /* Bad file descriptor */ #define ECHILD 10 /* No child processes */ #define EDEADLK 11 /* Resource deadlock avoided */ 6 Error Handling common mistake for displaying errno from Linux errno man page: 7 Error Handling Description of the perror () system call. -

Filesystems HOWTO Filesystems HOWTO Table of Contents Filesystems HOWTO
Filesystems HOWTO Filesystems HOWTO Table of Contents Filesystems HOWTO..........................................................................................................................................1 Martin Hinner < [email protected]>, http://martin.hinner.info............................................................1 1. Introduction..........................................................................................................................................1 2. Volumes...............................................................................................................................................1 3. DOS FAT 12/16/32, VFAT.................................................................................................................2 4. High Performance FileSystem (HPFS)................................................................................................2 5. New Technology FileSystem (NTFS).................................................................................................2 6. Extended filesystems (Ext, Ext2, Ext3)...............................................................................................2 7. Macintosh Hierarchical Filesystem − HFS..........................................................................................3 8. ISO 9660 − CD−ROM filesystem.......................................................................................................3 9. Other filesystems.................................................................................................................................3 -
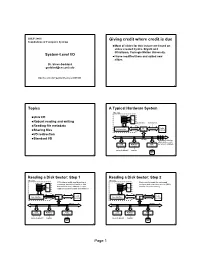
System-Level I/O I Have Modified Them and Added New Slides
JDEP 284H Giving credit where credit is due Foundations of Computer Systems Most of slides for this lecture are based on slides created by Drs. Bryant and O’Hallaron, Carnegie Mellon University. System-Level I/O I have modified them and added new slides. Dr. Steve Goddard [email protected] http://cse.unl.edu/~goddard/Courses/JDEP284 2 Topics A Typical Hardware System CPU chip register file Unix I/O ALU Robust reading and writing system bus memory bus Reading file metadata I/O main bus interface Sharing files bridge memory I/O redirection Standard I/O I/O bus Expansion slots for other devices such USB graphics disk as network adapters. controller adapter controller mouse keyboard monitor disk 3 4 Reading a Disk Sector: Step 1 Reading a Disk Sector: Step 2 CPU chip CPU chip CPU initiates a disk read by writing a Disk controller reads the sector and register file command, logical block number, and register file performs a direct memory access (DMA) destination memory address to a port transfer into main memory. ALU ALU (address) associated with disk controller. main main bus interface bus interface memory memory I/O bus I/O bus USB graphics disk USB graphics disk controller adapter controller controller adapter controller mouse keyboard monitor mouse keyboard monitor disk disk 5 6 Page 1 Reading a Disk Sector: Step 3 Unix Files CPU chip When the DMA transfer completes, the register file disk controller notifies the CPU with an A Unix file is a sequence of m bytes: interrupt (i.e., asserts a special “interrupt” B , B , ... -

System Programming Processes, Memory and Communication
System Programming Processes, memory and communication Thomas Ropars [email protected] 2021 1 The slides are available at: https://m1-mosig-os.gitlab.io/ 2 References Main references: • Advanced Programming in the Unix Environment by R. Stevens • The Linux Programming Interface by M. Kerrish • Operating Systems: Three Easy Pieces by R. Arpaci-Dusseau and A. Arpaci-Dusseau (introduction chapter) • Computer Systems: A Programmer's Perspective by R. Bryant and D. O'Hallaron The content of these lectures is inspired by: • The lecture notes of Prof. J.F. Mehaut. • The lecture notes of R. Lachaize. 3 Agenda What is an operating system? Unix File System The Shell Processes Inputs/Outputs 4 Agenda What is an operating system? Unix File System The Shell Processes Inputs/Outputs 5 Purpose of an operating system Operations run by a program: • Executing instructions • Reading/writing to memory • Reading/writing files • Accessing devices The operating system is here to make it easier to write and run programs, and to manage resources. 6 What is an operating system? Figure by R. Bryant and D. O'Hallaron The operating system is a layer of software interposed between the application program and the hardware Application programs Software Operating system Processor Main memory I/O devices Hardware Two main roles: • Virtualization • Resource management 7 Virtualization Transforms the physical resources into virtualized resources: • It hides the low-level interface of the hardware and provides higher-level abstractions: I easy to use I more general (hides the differences between different hardwares) I powerful I prevents programs from misusing the hardware It provides an API (Application Programming Interface) that allows user programs to interact with OS services: • A set of libraries • System calls 8 Resource management The OS allows several programs to run on the machine at the same time. -

GNU Findutils Finding Files Version 4.8.0, 7 January 2021
GNU Findutils Finding files version 4.8.0, 7 January 2021 by David MacKenzie and James Youngman This manual documents version 4.8.0 of the GNU utilities for finding files that match certain criteria and performing various operations on them. Copyright c 1994{2021 Free Software Foundation, Inc. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation License, Version 1.3 or any later version published by the Free Software Foundation; with no Invariant Sections, no Front-Cover Texts, and no Back-Cover Texts. A copy of the license is included in the section entitled \GNU Free Documentation License". i Table of Contents 1 Introduction ::::::::::::::::::::::::::::::::::::: 1 1.1 Scope :::::::::::::::::::::::::::::::::::::::::::::::::::::::::: 1 1.2 Overview ::::::::::::::::::::::::::::::::::::::::::::::::::::::: 2 2 Finding Files ::::::::::::::::::::::::::::::::::::: 4 2.1 find Expressions ::::::::::::::::::::::::::::::::::::::::::::::: 4 2.2 Name :::::::::::::::::::::::::::::::::::::::::::::::::::::::::: 4 2.2.1 Base Name Patterns ::::::::::::::::::::::::::::::::::::::: 5 2.2.2 Full Name Patterns :::::::::::::::::::::::::::::::::::::::: 5 2.2.3 Fast Full Name Search ::::::::::::::::::::::::::::::::::::: 7 2.2.4 Shell Pattern Matching :::::::::::::::::::::::::::::::::::: 8 2.3 Links ::::::::::::::::::::::::::::::::::::::::::::::::::::::::::: 8 2.3.1 Symbolic Links :::::::::::::::::::::::::::::::::::::::::::: 8 2.3.2 Hard Links ::::::::::::::::::::::::::::::::::::::::::::::: 10 2.4 Time -

GNU Find Utility Manual
Finding Files Edition 4.5.11-git, for GNU find version 4.5.11-git 17 November 2012 by David MacKenzie and James Youngman This file documents the GNU utilities for finding files that match certain criteria and per- forming various operations on them. Copyright c 1994, 1996, 1998, 2000, 2001, 2003, 2004, 2005, 2006, 2007, 2008, 2009, 2010, 2011 Free Software Foundation, Inc. Permission is granted to copy, distribute and/or modify this document under the terms of the GNU Free Documentation License, Version 1.3 or any later version published by the Free Software Foundation; with no Invariant Sections, with no Front-Cover Texts, and with no Back-Cover Texts. A copy of the license is included in the section entitled “GNU Free Documentation License”. i Table of Contents 1 Introduction..................................... 1 1.1 Scope .......................................................... 1 1.2 Overview ...................................................... 2 1.3 find Expressions ............................................... 3 2 Finding Files .................................... 5 2.1 Name .......................................................... 5 2.1.1 Base Name Patterns ....................................... 5 2.1.2 Full Name Patterns........................................ 5 2.1.3 Fast Full Name Search..................................... 7 2.1.4 Shell Pattern Matching .................................... 8 2.2 Links .......................................................... 9 2.2.1 Symbolic Links ........................................... -

CIS18A: Introduction to Linux/Unix CLASSROOM ATC
http://voyager.deanza.fhda.edu/~lalitha/cis18a/cis18a.html CIS18A: Introduction to Linux/Unix CLASSROOM ATC 204 WINTER 2015 : Section INFO: 00444 CIS -018A-61Y College academic Calendar: Winter 2015 http://deanza.fhda.edu/calendar/winterdates.html Instructor Information Name: Lalitha Krishnamurthy Office Hours: Not applicable ie None Email: krishnamurthylalitha at fhda dot edu Online Lab Hours: Wednesdays 845PM-10PM Lecture timings for CIS18A : M/W 6 PM - 750 PM Course Description This course is designed to discuss Linux/Unix Operating environment and its features. Linux/Unix commands, file structure, Regular expressions, shell features will be discussed Introduction to the features of the UNIX/LINUX operating system including text editing, text file manipulation, electronic mail, Internet utilities, directory structures, input/output handling, and shell features are part of the course curriculum CIS18A Student Learning Outcome Statements (SLO) Use the Unix/Linux Operating System utilities and shell features for basic file manipulation, networking, and communication. Course Objectives Edit text using the vi editor Maintain file and directory system Establish security and file permission, Perform basic file maintenance and use information utilities Utilize the shells Run shell commands, Implement quoting rules Communicate with email and communication utilities Apply filters and use implement basic regular expression Use basic utilities to explore system data, user data, and common tasks: exit, passwd, who, whoami, finger, w, tty, stty, uname, clear, -

Part Workbook 5. the Linux Filesystem Table of Contents
Part Workbook 5. The Linux Filesystem Table of Contents 1. File Details ................................................................................................................... 5 Discussion .............................................................................................................. 5 How Linux Save Files ...................................................................................... 5 What's in an inode? .......................................................................................... 6 Viewing inode information with the stat command ................................................. 9 Viewing inode information with the ls command .................................................. 10 Identifying Files by Inode ................................................................................ 10 Examples .............................................................................................................. 11 Example 1. Comparing file sizes with ls -s and ls -l ............................................... 11 Example 2. Listing files, sorted by modify time ..................................................... 11 Example 3. Decorating listings with ls -F ............................................................. 12 Online Exercises .................................................................................................... 13 Online Exercise 1. Viewing file metadata ............................................................. 13 Specification ......................................................................................... -

What Is Unix?
Unix vs. Linux • Age – Unix: born in 1970 at AT&T/Bell Labs – Linux: born in 1992 in Helsinki, Finland • Sun, IBM, HP are the 3 largest vendors of Unix – These Unix flavors all run on custom hardware • Linux is FREE! (Speech vs. Beer) – Linux was written for Intel/x86, but runs on many platforms – Reference by U of Washington CS department The Command Prompt • Commands are the way to “do things” in Unix • A command consists of a command name and options called “flags” • Commands are typed at the command prompt • In Unix, everything (including commands) is case-sensitive [prompt]$ <command> <flags> <args> fiji:~$ ls –l -a unix-tutorial (Optional) arguments Command Prompt Command (Optional) flags Note: Many Unix commands will print a message only if something went wrong. Be careful with rm and mv. Two Basic Commands • The most useful commands you’ll ever learn: – man (short for “manual”) – info • They help you find information about other commands – man <cmd> or info <cmd> retrieves detailed information about <cmd> – man –k <keyword> searches the man page summaries (faster, and will probably give better results) – Or use flag –-help to find the manual fiji:/u15/awong$ man –k password passwd (5) - password file xlock (1) - Locks the local X display until a password is entered fiji:/u15/awong$ passwd Two Basic Commands (info) • Info, as opposed to man, is category based • Documents are hyperlinked – info <cmd> retrieves detailed information about <cmd> – info by itself will give instructions on its usage – Type q to quit. – Type h for help.