US Department of Commerce Noaa NATIONAL OCEANIC and ATMOSPHERIC ADMINISTRATION
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Laboratory Glassware N Edition No
Laboratory Glassware n Edition No. 2 n Index Introduction 3 Ground joint glassware 13 Volumetric glassware 53 General laboratory glassware 65 Alphabetical index 76 Índice alfabético 77 Index Reference index 78 [email protected] Scharlau has been in the scientific glassware business for over 15 years Until now Scharlab S.L. had limited its sales to the Spanish market. However, now, coinciding with the inauguration of the new workshop next to our warehouse in Sentmenat, we are ready to export our scientific glassware to other countries. Standard and made to order Products for which there is regular demand are produced in larger Scharlau glassware quantities and then stocked for almost immediate supply. Other products are either manufactured directly from glass tubing or are constructed from a number of semi-finished products. Quality Even today, scientific glassblowing remains a highly skilled hand craft and the quality of glassware depends on the skill of each blower. Careful selection of the raw glass ensures that our final products are free from imperfections such as air lines, scratches and stones. You will be able to judge for yourself the workmanship of our glassware products. Safety All our glassware is annealed and made stress free to avoid breakage. Fax: +34 93 715 67 25 Scharlab The Lab Sourcing Group 3 www.scharlab.com Glassware Scharlau glassware is made from borosilicate glass that meets the specifications of the following standards: BS ISO 3585, DIN 12217 Type 3.3 Borosilicate glass ASTM E-438 Type 1 Class A Borosilicate glass US Pharmacopoeia Type 1 Borosilicate glass European Pharmacopoeia Type 1 Glass The typical chemical composition of our borosilicate glass is as follows: O Si 2 81% B2O3 13% Na2O 4% Al2O3 2% Glass is an inorganic substance that on cooling becomes rigid without crystallising and therefore it has no melting point as such. -

Process-Manager-User-Guide-En.Pdf
SAP Signavio Process Manager User Guide 15.6 Our guides available at documentation.signavio.com contain video examples, these examples are not included in the PDF versions. For more information, please contact the SAP Signavio doc- umentation team. Version 15.6 2 Contents 0.1 Welcome to the SAP Signavio Process Manager user guide 9 0.2 Signing up 9 0.2.1 Create your Business Transformation Suite account 10 0.2.2 Supported browsers 10 0.3 Log in to the Business Transformation Suite 11 0.3.1 Log in with your account credentials 11 0.3.2 Log in using a shared link 12 0.3.3 Log in to the on-premises solution 12 0.3.4 Next steps 12 0.4 Getting started 12 0.4.1 The explorer 13 0.4.2 The editor 13 0.4.3 QuickModel 13 0.4.4 The dictionary 13 0.4.5 SAP Signavio Process Collaboration Hub 13 0.4.6 The Diagram and Revision Comparison Tool 14 0.4.7 BPMN and DMN Simulation 14 0.4.8 Support 14 0.4.9 Next steps 14 0.4.10 What kind of SAP Signavio user am I 14 0.4.11 Explorer overview 16 0.4.12 The Explorer view 17 0.4.13 The Explorer menu 20 0.4.14 Personal profile settings 23 0.4.15 Working with folders and diagrams 26 0.4.16 Viewing diagram details 33 0.4.17 Today's top tips 34 0.4.18 The BPM Academic Initiative 35 0.4.19 Frequently asked questions 36 0.5 Modeling 37 0.5.1 Create a diagram 38 Version 15.6 3 0.5.2 Open and save diagrams 40 0.5.3 Editor toolbar and keyboard shortcuts 41 0.5.4 Add and connect elements 45 0.5.5 Move and change elements 48 0.5.6 Format diagrams 52 0.5.7 Work with modeling conventions 54 0.5.8 The dictionary 56 0.5.9 Process -
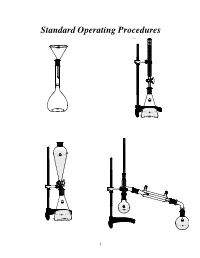
Standard Operating Procedures
Standard Operating Procedures 1 Standard Operating Procedures OVERVIEW In the following laboratory exercises you will be introduced to some of the glassware and tech- niques used by chemists to isolate components from natural or synthetic mixtures and to purify the individual compounds and characterize them by determining some of their physical proper- ties. While working collaboratively with your group members you will become acquainted with: a) Volumetric glassware b) Liquid-liquid extraction apparatus c) Distillation apparatus OBJECTIVES After finishing these sessions and reporting your results to your mentor, you should be able to: • Prepare solutions of exact concentrations • Separate liquid-liquid mixtures • Purify compounds by recrystallization • Separate mixtures by simple and fractional distillation 2 EXPERIMENT 1 Glassware Calibration, Primary and Secondary Standards, and Manual Titrations PART 1. Volumetric Glassware Calibration Volumetric glassware is used to either contain or deliver liquids at a specified temperature. Glassware manufacturers indicate this by inscribing on the volumetric ware the initials TC (to contain) or TD (to deliver) along with the calibration temperature, which is usually 20°C1. Volumetric glassware must be scrupulously clean before use. The presence of streaks or droplets is an indication of the presence of a grease film. To eliminate grease from glassware, scrub with detergent solution, rinse with tap water, and finally rinse with a small portion of distilled water. Volumetric flasks (TC) A volumetric flask has a large round bottom with only one graduation mark positioned on the long narrow neck. Graduation Mark Stopper The position of the mark facilitates the accurate and precise reading of the meniscus. If the flask is used to prepare a solution starting with a solid compound, add small amounts of sol- vent until the entire solid dissolves. -

Environmental Protection Agency Pt. 63, App. A
Environmental Protection Agency Pt. 63, App. A APPENDIX A TO PART 63—TEST METHODS posed as an alternative test method to meet an applicable requirement or in the absence METHOD 301—FIELD VALIDATION OF POLLUT- of a validated method. Additionally, the val- ANT MEASUREMENT METHODS FROM VARIOUS idation procedures of Method 301 are appro- WASTE MEDIA priate for demonstration of the suitability of alternative test methods under 40 CFR parts USING METHOD 301 59, 60, and 61. If, under 40 CFR part 63 or 60, 1.0 What is the purpose of Method 301? you choose to propose a validation method other than Method 301, you must submit and 2.0 What approval must I have to use Method obtain the Administrator’s approval for the 301? candidate validation method. 3.0 What does Method 301 include? 2.0 What approval must I have to use Method 301? 4.0 How do I perform Method 301? If you want to use a candidate test method REFERENCE MATERIALS to meet requirements in a subpart of 40 CFR part 59, 60, 61, 63, or 65, you must also request 5.0 What reference materials must I use? approval to use the candidate test method according to the procedures in Section 16 of SAMPLING PROCEDURES this method and the appropriate section of 6.0 What sampling procedures must I use? the part (§ 59.104, § 59.406, § 60.8(b), § 61.13(h)(1)(ii), § 63.7(f), or § 65.158(a)(2)(iii)). 7.0 How do I ensure sample stability? You must receive the Administrator’s writ- ten approval to use the candidate test meth- DETERMINATION OF BIAS AND PRECISION od before you use the candidate test method to meet the applicable federal requirements. -

Winter Antiques & Fine Art Auction
Winter Antiques Winter & Art Auction Fine Wednesday 27, Thursday 28 & Friday 29 November 2019 Thursday 28 & Friday 29 November 27, Wednesday Winter Antiques & Fine Art Auction Wednesday 27, Thursday 28 & Friday 29 November 2019 Chris Ewbank, FRICS, ASFAV Andrew Ewbank, BA, ASFAV John Snape, BA, ASFAV Alastair McCrea, MA Senior partner Partner Partner Partner [email protected] [email protected] [email protected] [email protected] Andrew Delve, MA, ASFAV Tim Duggan, ASFAV Andrea Machen, Cert GA Emily Angus, BA, FGA Partner Partner Jewellery Specialist Gemmologist [email protected] [email protected] [email protected] [email protected] Front cover: Lot 1137 Inside front cover: Lots 1, 2099 & 2036 Back cover: Lot 385 WINTER ANTIQUES & FINE ART AUCTION Surrey & Hampshire’s Premier Auctioneers & Valuers Winter Antiques & Fine Art Auction Jewellery & Costume Jewellery, Watches, Coins, Silver Plate, Silver, Fine Art, Ceramics & Glass, Collectables & Militaria, Books & Maps, Works of Art & Tea Caddies, Clocks, Antique Furniture and Persian Rugs SALE: Wednesday 27, Thursday 28 & Friday 29 November 2019 from 9.30am VIEWING: Saturday 23 November 10am - 2pm Monday 25 November 9am - 5pm Tuesday 26 November 9am - 7pm Days of Sale For the fully illustrated catalogue, to leave commission bids, and to register for Ewbank’s Live Internet Bidding please visit our website: www.ewbankauctions.co.uk The Burnt Common Auction Rooms London Road, Send, Surrey GU23 7LN Tel +44 (0)1483 223101 E-mail: [email protected] Buyer’s Premium at 28.8% inclusive of VAT, is payable on every lot in this sale. -

Q & a – Accuracy and Precision
Food Analysis – FScN 4312W Laboratory: Assessment of Accuracy and Precision Key to Questions 1. Theoretically, how are standard deviation, coefficient of variation, mean, percent relative error, and 95% confidence interval affected by: (1) more replicates, and (2) a larger size of measurement? Was this evident in looking at the actual results obtained using the volumetric pipettes and the buret, with n = 3 vs n = 6, and with 1mL vs 10mL? Ans: (1) As the sample size increases (more replicates) - Calculated mean approaches the true mean - Standard deviation is inversely proportional to the square root of sample size, hence it decreases - CV decreases, as standard deviation approaches 0 for larger sample size - % Relative error decreases as calculated mean approaches true mean - 95% confidence interval narrows down (2) Larger size of measurement - Mean is close to true mean - Standard deviation might be larger due to larger size - CV decreases - % Relative error decreases, as calculated mean is close to true mean - 95% confidence interval narrows down Not all of the above assumptions were confirmed in this experiment according to the data observed. One reason could be that the sample size was very small (n = 3 and n = 6), to notice significant differences, another reason could be human error and variation from one person’s measuring technique to another. 2. Why are percent relative error and coefficient of variation used to compare the accuracy and precision, respectively, of the volumes from pipetting/dispensing 1 and 10mL with the volumetric pipettes and buret in parts 2 and 3, rather than simply the mean and standard deviation, respectively? Ans: Mean calculated from the readings gives the calculated mean, which may differ from the true mean. -

Product Manual CH-4434 Hölstein Phone +41 61 956 11 11 Fax +41 61 951 20 65 [email protected] Contents
Oris SA Ribigasse 1 Product Manual CH-4434 Hölstein Phone +41 61 956 11 11 Fax +41 61 951 20 65 [email protected] www.oris.ch Contents. 7 English Introduction . 9 Adjusting Oris watches to fit the wrist . 20 Watches with leather straps . 20 Starting Oris watches . 10 Watches with rubber straps . 20 Crown positions . 10 Watches with metal bracelets . 20 Standard crown . 10 Fine adjustment of folding clasps . 20 Screw-down crown . 10 Crown with Oris Quick Lock system (QLC) . 10 Notes . 22 Screw-down pushers . 10 Accuracy . 22 Automatic winding watches . 11 Chronometer . 22 Manual winding watches . 11 Water-resistance . 24 Use and maintenance . 24 Setting and operating Oris watches . 12 Date, day of the week and time . 12 Technical information and Setting the date . 12 summary tables . 26 Worldtimer . 12 Pictograms . 26 Worldtimer with 3rd time zone and compass . 13 Metals for cases and straps . 27 2nd time zone on outer rotating bezel . 14 PVD coatings . 27 2nd time zone indicator on inner rotating Sapphire crystal . 27 bezel with vertical crown . 14 Mineral glass . 28 2nd time zone with additional 24 hr hand . 14 Plexi glass . 28 2nd time zone with additional 24 hr hand and Luminescent dials and hands . 28 city markers on the rotating bezel . 14 Metal bracelets, leather and rubber straps . 28 Chronograph . 15 Lunar calendar . 29 Complication . 15 Time zones . 30 Regulator . 16 Movements . 30 Pointer calendar . 16 Alarm with automatic winding . 16 International guarantee for Oris watches . 32 Tachymeter scale – measuring speeds . 17 Telemeter scale – measuring distances . 17 Proof of ownership . -

TOP-014, Technical Operating Procedure for Spectrophotometric
CENTER FOR NUCLEAR WASTE Proc. T0P-014 REGULATORY ANALYSES Revision ° TECHNICAL OPERATING PROCEDURE Page I of 11 Title TECHNICAL OPERATING PROCEDURE FOR SPECTROPHOTOMETRIC DETERMINATION OF SILICA EFFECTIVITY AND APPROVAL Revision n of this procedure became effective on 03/04/91 This procedure consists of the pages and changes listed below. Page No. Change Date Effective ALL 03/04/91 Superseaes Procedure No. None Approvals Written By Technical Revie / CENTER FOR NUCLEAR WASTE Proc.TOP-014 REGULATORY ANALYSES Revision _ TECHNICAL OPERATING PROCEDURE Page 2 of 1il TOP-014 TECHNICAL OPERATING PROCEDURE FOR SPECTROPHOTOMETRIC DETERMINATION OF SILICA 1. PURPOSE The purpose of this procedure is to describe a general method for the spectrophotometric determination of silica (Si02) in aqueous solutions. This procedure will be used for aqueous solutions generated by geochemical experiments involving silicate minerals (e.g., zeolites and feldspars) but may also be used for analyses of water samples collected from laboratory experiments or in the field. This procedure implements the requirements of CQAM Section 3. 2. APPLICABLE DOCUMENT The following document forms a part of this procedure, as applicable: Operator's Manual for Milton Roy Spectronic 1201 Spectrophotometer (1988) 3. RESPONSIBILITY (1) The Geosciences Element Manager shall be responsible for the development and maintainance of this procedure. (2) The cognizant Principal Investigator shall be responsible for the implementation of this procedure. (3) Personnel performing tasks described in this procedure are responsible for complying with its requirements. 4. EQUIPMENT (1) Milton Roy Spectronic 1201 Spectrophotometer with microprocessor-controlled functions and features including a linear curve fit test mode or equivalent. -

Standardization Framework for Sustainability from Circular Economy 4.0
sustainability Article Standardization Framework for Sustainability from Circular Economy 4.0 María Jesús Ávila-Gutiérrez * , Alejandro Martín-Gómez, Francisco Aguayo-González and Antonio Córdoba-Roldán Design Engineering Dept, University of Seville, Escuela Politécnica Superior, Virgen de África 7, 41011 Seville, Spain; [email protected] (A.M.-G.); [email protected] (F.A.-G.); [email protected] (A.C.-R.) * Correspondence: [email protected] Received: 8 October 2019; Accepted: 15 November 2019; Published: 18 November 2019 Abstract: The circular economy (CE) is widely known as a way to implement and achieve sustainability, mainly due to its contribution towards the separation of biological and technical nutrients under cyclic industrial metabolism. The incorporation of the principles of the CE in the links of the value chain of the various sectors of the economy strives to ensure circularity, safety, and efficiency. The framework proposed is aligned with the goals of the 2030 Agenda for Sustainable Development regarding the orientation towards the mitigation and regeneration of the metabolic rift by considering a double perspective. Firstly, it strives to conceptualize the CE as a paradigm of sustainability. Its principles are established, and its techniques and tools are organized into two frameworks oriented towards causes (cradle to cradle) and effects (life cycle assessment), and these are structured under the three pillars of sustainability, for their projection within the proposed framework. Secondly, a framework is established to facilitate the implementation of the CE with the use of standards, which constitute the requirements, tools, and indicators to control each life cycle phase, and of key enabling technologies (KETs) that add circular value 4.0 to the socio-ecological transition. -

Purchasing Policies PDF Opens in New Window
Revised June 4, 2020 Table of Contents SECTION 1 – LCCC BOARD OF TRUSTEES Bylaws Lehigh Carbon Community College Board of Trustees .............................................................................................. 1-1 Article I Definitions.............................................................................................................................................. 1-1 Article II Authorization ......................................................................................................................................... 1-1 Article III Board of Trustees Membership .............................................................................................................. 1-1 Article IV Officers .................................................................................................................................................. 1-2 Article V Election of Officers ................................................................................................................................ 1-2 Article VI Meetings ................................................................................................................................................ 1-2 Article VII Committees ........................................................................................................................................... 1-3 Article VIII Order of Business ............................................................................................................................. 1-3 Article -

List of Equipment
Mfg & Exporters: All types of Turnkey Projects, , Beverages, Mineral Water plant & OEM Spares List Of Equipment SR.NO Equipment Qty 1. Autoclave 1 Vertical Complete Heavy gauze with pressure gauze, Safety Valve, Stream Release Valve Size 30 X 30 cm 2. Digital Hot Air Ageing oven 1 Size 12” x 12” x 12” made of MS powder coated. Make- HB 3. Digital pH System : 1 Make- HB 4. Digital Incubator Bacteriological 2 12” X 12” X 12” made of MS powder coated. Make- HB 5. Water bath double walled 1 inner chamber of SS outer M.S powder coated with 6 hole with thermostatic control Make- HB 6. INNOCULATION CHAMBER, M.S POWDER COATED WITH 1 UV & 1 FLOROCENT LIGHT INBUILT 1 Make- HB 7. Automatic Hot Plate with 8 inch dia with Thermostat 1 Make-HB 8. Digital Spectrophotometer 1 Make- HB 9. Digital Turbidity meter 1 Make- HB 10. Digital Colony Counter 1 Make- HB 11. S.S. Filter holder 47 mm 1 (complete S.S) 12. Microscope student 1 Make-Labron 13. Magnetic Stirrer with Hot Plate with 1 lit capacity Top made of SS with Teflon Bed 1 Make- HB 14. Centrifuge ( 4 holders) 1 Make- HB 15. Single pan Balance ( pocket type) max: 0-500 gm, L.C: 0.1 gm 1 16. High pressure vacuum Pump 1 17. BOD Incubators inner complete S.S outer MS powder coated.( heavy) 2 Make – HB 18. Heating Mental 1 lit capacity 1 Make- HB 19. S.S distillation Unit 1 Address: 140, Tribhuvan Estate opp Road No 11 Kathwada GIDC Contact: +91 9726943972 Email: [email protected] Mfg & Exporters: All types of Turnkey Projects, , Beverages, Mineral Water plant & OEM Spares List Of Chemicals Sr. -

Auction House KANERZ ART PUBLIC SALE PRESTIGE ITEMS
Auction House KANERZ ART PUBLIC SALE PRESTIGE ITEMS Watches, Jewelry, Goldsmithing, Asian & African Art, Paintings, Fashion, Books including Law & Medicine, Art of Living, Furniture, Sculptures, Bronzes... Sunday, May 24th, 2020, at 2 PM Part I (10 AM to 12 AM) Jewelry, Watches, Lifestyle, Books, Medals, Asian Art… Part II (2 PM to 6 PM) African Art, Silverware, Bronze, Furniture, Paintings, Porcelain... The sale is broadcast live with possibility of online auction on Exhibition and sale in the room will take place under sanitary conditions in accordance with government recommendations (barrier gestures & limitation of the number of people in the room at the same time). In order to respect the distances on the day of the sale, only 20 people will be admitted upon registration. Exhibition of the lots: Thursday May 21 & Friday May 22, from 9 AM to 6 PM without interruption, Saturday May 23, from 10 AM to 2 PM. Sales Expert for Asian Items: PHILIPPE DELALANDE EXPERTISE 23 rue Lemercier 75017 Paris, France Tel: +33 (0) 6 83 11 24 71 Email: [email protected] All the lots are in photo on the site of the House of sale: www.encheres-luxembourg.lu Contact : [email protected] GSM : (+352) 621.612.226 Place of the sale: 35 Rue Kennedy, L-7333 Steinsel Free Parking Photos, pre-orders and general conditions of sale can be found on the website of the auction house KANERZ ART at www.encheres-luxembourg.lu. The sale is broadcast live with possibility of online and live auction on www.auction.fr Maison de Ventes KANERZ ART The visits will be an opportunity for buyers to get a clear idea of the desired objects beyond the formal description of the catalog.