〈541〉 Titrimetry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The International Pharmacopoeia
The International Pharmacopoeia THIRD EDITION Pharmacopoea internationalis Editio tertia Volume 4 Tests, methods, and general requirements Quality specifications for pharmaceutical substances, excipients, and dosage forms World Health Organization Geneva 1994 WHO Library Cataloguing in Publication Data The International Pharmacopoeia.- 3rd ed. Contents: v. 4. Tests, methods, and general requirements 1. Drugs -analysis 2. Drugs -standards ISBN 92 4 154462 7 (NLM Classification: QV 25) The World Health Organization welcomes requests for permission to reproduce or translate its publica- tions, in part or in full. Applications and enquiries should be addressed to the Of£ice of Publications, World Health Organization, Geneva, Switzerland, which will be glad to provide the latest information on any changes made to the text, plans for new editions, and reprints and translations already available. O World Health Organization, 1994 Publications of the World Health Organization enjoy copyright protection in accordance with the provi- sions of Protocol 2 of the Universal Copyright Convention. All rights reserved. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. The mention of specific companies or of certain manufacturers' products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distin- guished by initial capital letters. -

Biomolecules - Biochimiques - Standards
BIOMOLECULES - BIOCHIMIQUES - STANDARDS H Tél. : 33 (0)4 70 03 88 55 I Hotline : 33 (0)4 70 03 73 06 I E-mail : [email protected] I Fax : 33 (0)4 70 03 82 60 BIOMOLECULES - BIOCHIMIQUES - STANDARDS BIOMOLECULES - BIOCHIMIQUES - STANDARDS Standards de Dioxyde de Carbone H.93 Standards et réactif pour analyseurs de Chlorure H.94 Standards Couleur Hazen H.94 Standards Redox H.94 Standards d'Electrodes sélectives d'ions H.95 Solutions d'ajustage de force ionique H.95 Standards de chromatographie ionique H.96-H.97 Standards pour ICP H.98-H.106 Standards pour Photométrie de flamme H.107-H.108 Organiques H.109-H.123 Composés organiques volatils (COV) H.109 Biomolécules & Composés actifs H.2-H.36 Standards Mélangés de Composés Introduction H.2 Organiques Volatils H.110-H.111 Peptides H.3-H.4 Phénols H.112-H.113 Peptides bloquants pour anticorps H.3 Hydrocarbures aromatiques Peptides (bioactifs, aptamères, viraux…) H.3-H.4 polycycliques (HAP) H.114-H.115 Standards de Carbone Organique Total (COT) H.116 Protéines H.5-H.28 Pesticides H.117-H.118 Kinases H.5-H.13 Standards Lipidiques Avanti Polar Lipids H.119-H.121 Phosphatases Actives H.14 Standards Lipidiques Larodan H.122 Phosphodiestérases Actives H.14 Standards individuels - Contrôles - Histone Déacétylases Actives H.15 Calibrateurs pour hématologie H.123 Acetyltransférases Actives H.15 Acétyl/Methyltransférases Inactives H.15 Couleurs H.124-H.126 Autres enzymes H.16 Standards de couleurs H.124-H.126 Substrats Enzymatiques H.17-H.18 Spectrophotométrie H.127-H.130 Marqueurs CD H.19 Standards -
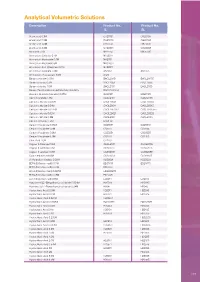
Analytical Volumetric Solutions
Analytical Volumetric Solutions Description Product No. Product No. 1L 5L Acetic acid 0.1M CH20101 CH20105 Acetic acid 0.5M CH20051 CH20055 Acetic acid 1.0M CH21001 CH21005 Acetic acid 2.0M CH22001 CH22005 Ammonia 0.1M NH20101 NH20105 Ammonium Chloride 0.1M NHCl011 Ammonium Hydroxide 0.5M NH2051 Ammonium Hydroxide 6M NH32601 Ammonium Iron (II)Sulphate 0.1M NHS2011 Ammonium sulphate 0.5M AS2051 AS2055 Ammonium Thiocyanate 1.0M AT21F Barium chloride 0.05M BACL20051 BACL20055 Barium chloride 0.5M BACL2051 BACL2055 Barium chloride 1.0M BACL2101 BACL2105 Barium Perchlorate 0.005M Alcoholic Solution BACLO200051 Bromine (Bromate/bromide) 0.05M BR20101 BR20105 Calcium acetate 1.0M CAAC2101 CAAC2105 Calcium chloride 0.005M CACL20051 CACL20055 Calcium chloride 0.01M CACL20011 CACL20015 Calcium chloride 0.0125M CACL2001251 CACL2001255 Calcium chloride 0.02M CACL20021 CACL20025 Calcium chloride 0.5M CACL2051 CACL2055 Calcium Chloride 1.0M CACL101 Cerium IV sulphate 0.05M CS20051 CS20055 Cerium IV sulphate 0.1M CS2011 CS2015 Cerium IV sulphate 0.2M CS20251 CS20255 Cerium IV sulphate 1.0M CS2101 CS2105 Citric Acid 1.0M CH2101 Copper II Chloride 0.5M CUCL2051 CUCL2055 Copper II sulphate 0.1M CUS02011 CUS02015 Copper II sulphate 0.5M CUS02051 CUS02055 Cupric solution 0.168M CU201681 CU201685 Di-Potassium Oxalate 0.05M KO20051 KO20055 EDTA (DiSodium salt) 0.01M ED20011 ED20015 EDTA (DiSodium salt) 0.02M ED20021 EDTA (DiSodium Salt) 0.027M EDB200271 EDTA (DiSodium salt) 0.05M ED20051 EDTA (DiSodium salt) 0.1M ED2011 ED2015 Hyamine 1622 -Benzethonium chloride -

Product Catalog
For a regularly updated list of pricing and availalble sizes, visit us at www.reagents.com. call: 800-732-8484 fax: 888-843-4384 email: [email protected] 2021 Cover.indd 1 4/26/21 7:55 PM Reagents is your One-Stop Shop Reagents has been a leading manufacturer and distributor of specialty chemicals, reagents, and analytical testing solutions for over 50 years. As Reagents continues to grow, we are constantly adding new brands to our selection. We’ve added a wide selection of lab supply and glassware brands with products that are tested to ASMT guidelines. These Reagents Specialty Reagents Provides: diverse additions allow us to provide a broad portfolio of world class brands manufactured utilizing the highest quality raw Chemicals and Solutions • ISO 17025 Accreditation / ISO 9001 Certified Manufacturing Facility materials meeting or exceeding specifications established by the American Chemical Society. In the laboratory or on the has been a leading • Meets or exceeds stringent manufacturing guidelines production floor, our diverse product portfolio allows consolidation of all of your scientific product needs. manufacturer and • Over 50,000 products available distributor of specialty • Full portfolio of world class brands chemicals, reagents and • High order fill rate within 48 hours on key items analytical testing solutions • Technical and applications support • Degreed and experienced chemical staff since 1969. • Competitive offering and pricing We utilize the highest • Tenured sales specialists with 20+ years of consultative experience quality raw materials to • GHS label compliance meet or exceed the • Many solutions certified traceable to NIST standard reference material specifications established by the American Chemical Custom Solutions: Society. -

FCC 10, Second Supplement the Following Index Is for Convenience and Informational Use Only and Shall Not Be Used for Interpretive Purposes
Index to FCC 10, Second Supplement The following Index is for convenience and informational use only and shall not be used for interpretive purposes. In addition to effective articles, this Index may also include items recently omitted from the FCC in the indicated Book or Supplement. The monographs and general tests and assay listed in this Index may reference other general test and assay specifications. The articles listed in this Index are not intended to be autonomous standards and should only be interpreted in the context of the entire FCC publication. For the most current version of the FCC please see the FCC Online. Second Supplement, FCC 10 Index / Allura Red AC / I-1 Index Titles of monographs are shown in the boldface type. A 2-Acetylpyrrole, 21 Alcohol, 90%, 1625 2-Acetyl Thiazole, 18 Alcohol, Absolute, 1624 Abbreviations, 7, 3779, 3827 Acetyl Valeryl, 608 Alcohol, Aldehyde-Free, 1625 Absolute Alcohol (Reagent), 5, 3777, Acetyl Value, 1510 Alcohol C-6, 626 3825 Achilleic Acid, 25 Alcohol C-8, 933 Acacia, 602 Acid (Reagent), 5, 3777, 3825 Alcohol C-9, 922 ªAccuracyº, Defined, 1641 Acid-Hydrolyzed Milk Protein, 22 Alcohol C-10, 390 Acesulfame K, 9 Acid-Hydrolyzed Proteins, 22 Alcohol C-11, 1328 Acesulfame Potassium, 9 Acid Calcium Phosphate, 240 Alcohol C-12, 738 Acetal, 10 Acid Hydrolysates of Proteins, 22 Alcohol C-16, 614 Acetaldehyde, 11 Acidic Sodium Aluminum Phosphate, Alcohol Content of Ethyl Oxyhydrate Acetaldehyde Diethyl Acetal, 10 1148 Flavor Chemicals (Other than Acetaldehyde Test Paper, 1636 Acidified Sodium Chlorite -

ABSTRACT WILLIAMS, TOVA N. Approaches
ABSTRACT WILLIAMS, TOVA N. Approaches to the Design of Sustainable Permanent Hair Dyes (Under the direction of Harold S. Freeman). Interest in designing sustainable permanent hair dyes derives from the toxicological concerns associated with certain commercial dyes, in particular the moderate to strong/extreme skin sensitization potential of certain precursors used to develop the dyes. While work has been undertaken to help address toxicological concerns of hair dyes, alternatives having commercial success are based on the conventional permanent hair dye coloration process and still may pose health problems. The conventional process involves the oxidation of small and essentially colorless precursors (e.g., p-phenylenediamine and resorcinol) within the hair fiber that couple to build oligomeric indo dyes that are difficult to desorb. The resultant depth of shade achieved is superior that of other hair dyes and provides a high degree of gray coverage. In fact, these dyes dominate the commercial realm and stimulate the multibillion-dollar global hair dye market for the millions of consumers (men and women) who use them. Approaches to the design of sustainable (less toxic) permanent hair dyes lie at the heart of the present study. In Part 1 of this study, two types of keratin films (opaque and translucent) were characterized for their potential as screening tools for predicting the efficacy of potential hair dyes using the hair dye, C.I. Acid Orange 7. Translucent keratin films, which were found to be less porous than opaque films and more like hair in this regard, were deemed better tools for predicting the efficacy of potential hair dyes on hair. -

Fcc-10-Index.Pdf
FCC 10 Index / Allyl Beta-phenylacrylate / I-1 Index Titles of monographs are shown in the boldface type. A 2-Acetyl Thiazole, 18 Alcohol C-6, 626 Acetyl Valeryl, 608 Alcohol C-8, 933 Abbreviations, 7 Acetyl Value, 1510 Alcohol C-9, 922 Absolute Alcohol (Reagent), 5 Achilleic Acid, 25 Alcohol C-10, 390 Acacia, 602 Acid (Reagent), 5 Alcohol C-11, 1328 ªAccuracyº, Defined, 1641 Acid-Hydrolyzed Milk Protein, 22 Alcohol C-12, 738 Acesulfame K, 9 Acid-Hydrolyzed Proteins, 22 Alcohol C-16, 614 Acesulfame Potassium, 9 Acid Calcium Phosphate, 240 Alcohol Content of Ethyl Oxyhydrate Acetal, 10 Acid Hydrolysates of Proteins, 22 Flavor Chemicals (Other than Acetaldehyde, 11 Acidic Sodium Aluminum Phosphate, Essential Oils), 1547 Acetaldehyde Diethyl Acetal, 10 1148 Alcohol, Diluted, 1624 Acetaldehyde Test Paper, 1636 Acidified Sodium Chlorite Alcoholic Potassium Hydroxide TS, Acetals (Essential Oils and Flavors), Solutions, 24 1625 1505 Acidity Determination by Iodometric Alcoholometric Table, 1729 Acetanisole, 12 Method, 1547 Aldehyde C-6, 616 Acetate C-10, 389 Acid Magnesium Phosphate, 791 Aldehyde C-7, 607 Acetate Identification Test, 1427 Acid Number (Rosins and Related Aldehyde C-8, 926 Aceteugenol, 500 Substances), 1529 Aldehyde C-9, 916 Acetic Acid Furfurylester, 543 Acid Phosphatase Activity, 1474 Aldehyde C-10, 386 Acetic Acid, Glacial, 12 Acid Phthalate Buffer, 1623 Aldehyde C-11 Undecyclic, 1323 Acetic Acid TS, Diluted, 1624 Acid Sodium Pyrophosphate, 1145 Aldehyde C-11 Undecylenic, 1326 Acetic Acid TS, Strong, 1624 Acid Trisodium Pyrophosphate, -

D Y E S C L a S S I F I E I B Y I N T E R M E D I a T E ! Dyes Tabularly Arranged Under Each Intermediate, with Statistical
DYES CLASSIFIEI BY INTERMEDIATE! Dyes tabularly arranged under each intermediate, with statistical and other data for both dyes and intermediates. Glossary of Dye and Intermediate names alphabetically arranged. BY R. XORRIS SHREVE Consulting Chemist EN" COLLABORATION WITH WARREN N. WATSON AND A. R. WILLIS Chemists, U. S. Tariff Commission BOOK DEPARTMENT The CHEMICAL CATALOG CCttBPANY, Inc. ONE MADISON AVKNUJS, NEW YORK, TJ. 8. A. COPYRIGHT, 1922, BY The CHEMICAL CATALOG COMPANY, Inc. All Rights Reserved Press of J. J. Little & Ives Company New York, U. S. A. TABLE OF CONTEXTS PAGE PREFACE 3 ABBREVIATION'S 5 INTRODUCTION 7 PART I. INTERMEDIATES AND DYE TABLES ... 17 KEY TO PART I 18 INTERMEDIATES WITH DYE TABLES. THE ARRANGEMENT OF INTER- MEDL^TE NAMES 15 ALPHABETICAL AND INCLUDES CHEMICAL AND COMMON OR TRIVIAL- NAMES 19 FORMULA INDEX OF INTERMEDIATES 581 PAET II. DYE NAMES 587 GLOSSARY OF DYE NAMES 589 PAGE INDEX OF SCHULTZ NUMBERS FOR DYES 625 PREFACE Experience in the manufacture of dyes indicates that the proper viewpoint for a correct technical program is from the intermediate side. This is a direct corollary of the fact that the intermediates are the materials out of which dyes are fabricated. Furthermore, the tremen- dous complexity of the dye industry, the interrelationship of one dye to another or of one intermediate to another, as well as the relationship of dyes and intermediates to the whole organic chemical industry, all require that there be available tables showing the commercial dyes derived from each important intermediate. To give this is the prime object of this work. -

Excerpted USP-NF and FCC Standards: a Hand Sanitizer Resource
Not official text. Please refer to the currently official version of the applicable USP–NF or FCC standard for compliance purposes. Excerpted USP-NF and FCC Standards: A Hand Sanitizer Resource A collection of standards provided as a resource to assist with the challenges posed by COVID-19 Not official text. Please refer to the currently official version of the applicable USP–NF or FCC standard for compliance purposes. Published August 17, 2020 Not official text. Please refer to the currently official version of the applicable USP–NF or FCC standard for compliance purposes. CONTENTS USP Monographs…………………………………………………………………15 • Alcohol • Dehydrated Alcohol • Azeotropic Isopropyl Alcohol • Glycerin • Hydrogen Peroxide Concentrate • Hydrogen Peroxide Topical Solution • Isopropyl Alcohol • Purified Water Excipients………………………………………………………………………… 25 • USP and NF Excipients, Listed by Functional Category NF Monographs…………………………………………………………………. 37 • Denatonium Benzoate • Sucrose Octaacetate Reference Tables……………………………………………………………….. 39 • Alcoholometric Table Food Chemicals Codex……………………………………………………… 119 • Ethyl Alcohol • Glycerin • Hydrogen Peroxide • Isopropyl Alcohol Additional documentary standards are also included in this publication to support these highlighted standards. Published August 17, 2020 Not official text. Please refer to the currently official version of the applicable USP–NF or FCC standard for compliance purposes. Published August 17, 2020 Not official text. Please refer to the currently official version of the applicable USP–NF