Identification of Sapovirus GV. 2, Astrovirus VA3 and Novel
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

GB Virus C/Hepatitis G Virus Does Not Induce Expression of P44 Antigen in Chimpanzee Hepatocytes Yohko K
Jpn. J. Infect. Dis., 54, 2001 and skin were removed was purchased on the previous night ongln Of infection was not clear. The food service in a festival of the festival. It was left in the room temperature ovemight. as described in the present case does not requlre regulation The cooking started at 4 0'clock in the momlng by roastlng under the food hyglene law. This incident indicated the the bulk meat by rotation over a gas bumer. The meat was necessity of proper measures for food security in this type of covered by a large steel box during roastlng. It was cut into short temporary food service and regulatory measures in case small pleCeS and served at 10 o'clock in the momlng. Some of possible occurrences of food poISOnlng. noted that some portion of the meat was rare; i.e., it was insufficiently cooked. Laboratory and other epidemiologlCal data will be published ln the outbreak, a total 58 cases were counted. There were in Infectious Agents Surveillance Report, vol. 22 (June, 200 I ). 41 primary infections, 1 1 secondary infections including a We thank the clinical institutions, schools and other institu- case which took place in a nursery school, and 6 cases whose tions fわr their collaboration. Laboratory and Epidemiology Communications GB Virus C/Hepatitis G Virus Does Not Induce Expression of p44 Antigen in Chimpanzee Hepatocytes Yohko K. Shimizu*, Minako Hijikata and Hiroshi Yoshikural Department ofRespiratoyy Diseases, Research Institute, International Medical Center ofJapan, Toyama 1-21-1 Shinjuku-ku, Too,0 162-8655 and JNational Institute ofInfectious Diseases, Toyama 1-23-1, Shinjuku-ku, Tokyo 162-8640 Communicated by Hiroshi Ybshikura (Accepted June 1, 2001) The cytoplasmic antlgen, p44, was orlglnally discovered found chimpanzees whose sera were positive for GBV-C什IGV in hepatocytes of chimpanzees experimentally infected with RNA by RTIPCR. -

Pdf Available
Virology 554 (2021) 89–96 Contents lists available at ScienceDirect Virology journal homepage: www.elsevier.com/locate/virology Diverse cressdnaviruses and an anellovirus identifiedin the fecal samples of yellow-bellied marmots Anthony Khalifeh a, Daniel T. Blumstein b,**, Rafaela S. Fontenele a, Kara Schmidlin a, C´ecile Richet a, Simona Kraberger a, Arvind Varsani a,c,* a The Biodesign Center for Fundamental and Applied Microbiomics, School of Life Sciences, Center for Evolution and Medicine, Arizona State University, Tempe, AZ, 85287, USA b Department of Ecology & Evolutionary Biology, Institute of the Environment & Sustainability, University of California Los Angeles, Los Angeles, CA, 90095, USA c Structural Biology Research Unit, Department of Clinical Laboratory Sciences, University of Cape Town, 7925, Cape Town, South Africa ARTICLE INFO ABSTRACT Keywords: Over that last decade, coupling multiple strand displacement approaches with high throughput sequencing have Marmota flaviventer resulted in the identification of genomes of diverse groups of small circular DNA viruses. Using a similar Anelloviridae approach but with recovery of complete genomes by PCR, we identified a diverse group of single-stranded vi Genomoviridae ruses in yellow-bellied marmot (Marmota flaviventer) fecal samples. From 13 fecal samples we identified viruses Cressdnaviricota in the family Genomoviridae (n = 7) and Anelloviridae (n = 1), and several others that ware part of the larger Cressdnaviricota phylum but not within established families (n = 19). There were also circular DNA molecules identified (n = 4) that appear to encode one viral-like gene and have genomes of <1545 nts. This study gives a snapshot of viruses associated with marmots based on fecal sampling. -

First Description of Adenovirus, Enterovirus, Rotavirus and Torque
First description of Adenovirus, Enterovirus, Rotavirus and Torque teno virus in water samples collected from the Arroio Dilúvio, Porto Alegre, Brazil Vecchia, AD.a,b, Fleck, JD.a,b, Comerlato, J.c, Kluge, M.b, Bergamaschi, B.c, Da Silva, JVS.b, Da Luz, RB.b, Teixeira, TF.b, Garbinatto, GN.d, Oliveira, DV.d, Zanin, JG.d, Van der Sand, S.d, Frazzon, APG.d, Franco, AC.c, Roehe, PM.c,e and Spilki, FR.a,b* aPrograma de Pós-Graduação em Qualidade Ambiental, Universidade Feevale, CEP 93352-000, Novo Hamburgo, RS, Brazil bLaboratório de Microbiologia Molecular, Instituto de Ciências da Saúde, Universidade Feevale, CEP 93352-000, Novo Hamburgo, RS, Brazil cLaboratório de Virologia, Departamento de Microbiologia, Instituto de Ciências Básicas da Saúde, Universidade Federal do Rio Grande do Sul – UFRGS, Av. Sarmento Leite, 500, CEP 90050-170, Porto Alegre, RS, Brazil dDepartamento de Microbiologia, Instituto de Ciências Básicas da Saúde, Universidade Federal do Rio Grande do Sul – UFRGS, Av. Sarmento Leite, 500, CEP 90050-170, Porto Alegre, RS, Brazil eInstituto de Pesquisa Veterinária “Desidério Finamor” – IPVDF, Fundação Estadual de Pesquisa Agropecuária – FEPAGRO-Saúde Animal, Estrada do Conde, 6000, CEP 92990-000, Eldorado do Sul, RS, Brazil *e-mail: [email protected] Received May 11, 2011 – Accepted July 14, 2011 – Distributed May 31, 2012 (With 1 figure) Abstract Adenovirus (AdV), enterovirus (EV), genogroup A rotaviruses (GARV) and Torque teno virus (TTV) are non-enveloped viral agents excreted in feces and so may contaminate water bodies. In the present study, the molecular detection of these viruses was performed in samples of surface water collected from the Arroio Dilúvio, a waterstream that crosses the city of Porto Alegre, RS, Brazil, receiving great volumes of non-treated sewage from a large urban area. -

Prevalence and Clinical Significance of HGV/GBV-C Infection in Patients
Jpn. J. Infect. Dis., 59, 25-30, 2006 Original Article Prevalence and Clinical Significance of HGV/GBV-C Infection in Patients with Chronic Hepatitis B or C Jeng-Fu Yang1,2, Chia-Yen Dai2,3, Wan-Long Chuang2, Wen-Yi Lin1,2, Zu-Yau Lin2, Shinn-Cherng Chen2, Ming-Yuh Hsieh2, Liang-Yen Wang2, Jung-Fa Tsai2, Wen-Yu Chang2 and Ming-Lung Yu1,2* 1Department of Preventive Medicine and 2Department of Medicine, Kaohsiung Medical University Hospital, and 3Department of Occupational Medicine, Kaohsiung Municipal HsiaoKang Hospital, Kaohsiung, Taiwan (Received July 5, 2005. Accepted November 11, 2005) SUMMARY: Hepatitis G virus/GB virus-C (HGV/GBV-C) is a newly identified Flavivirus. Its clinical signifi- cance in chronic hepatitis B and C remains controversial. Infection with HGV/GBV-C was surveyed in 500 blood donors, 130 patients with chronic hepatitis B and 173 with hepatitis C, with chronic liver disease, cirrhosis, and/or hepatocellular carcinoma (HCC). HGV/GBV-C RNA was detected by reverse transcription-polymerase chain reaction. An antibody to HGV/GBV-C’s second envelope protein (anti-E2 Ab) was detected using an enzyme immunoassay. The prevalence of HGV/GBV-C RNA was 3.4% and the exposure rate 10.2% in blood donors. The prevalence of HGV/GBV-C RNA in patients with chronic hepatitis B and hepatitis C was 7.7 and 17.3%, respectively (P = 0.002). The prevalence of the HGV/GBV-C infection in hepatitis B carriers increased with the severity of chronic liver disease and risk of HCC. The age and duration of hepatitis B virus infection were the more important contributing factors. -

Non-Norovirus Viral Gastroenteritis Outbreaks Reported to the National Outbreak Reporting System, USA, 2009–2018 Claire P
Non-Norovirus Viral Gastroenteritis Outbreaks Reported to the National Outbreak Reporting System, USA, 2009–2018 Claire P. Mattison, Molly Dunn, Mary E. Wikswo, Anita Kambhampati, Laura Calderwood, Neha Balachandran, Eleanor Burnett, Aron J. Hall During 2009–2018, four adenovirus, 10 astrovirus, 123 The Study rotavirus, and 107 sapovirus gastroenteritis outbreaks NORS is a dynamic, voluntary outbreak reporting were reported to the US National Outbreak Reporting system. For each reported outbreak, health depart- System (annual median 30 outbreaks). Most were at- ments report the mode of transmission, number of tributable to person-to-person transmission in long-term confirmed and suspected cases, and aggregate epi- care facilities, daycares, and schools. Investigations of demiologic and demographic information as avail- norovirus-negative gastroenteritis outbreaks should in- able. NORS defines outbreaks as >2 cases of similar clude testing for these viruses. illness associated with a common exposure or epi- demiologic link (9). Health departments determine n the United States, ≈179 million cases of acute gas- reported outbreak etiologies on the basis of available troenteritis (AGE) occur annually (1). Norovirus is I laboratory, epidemiologic, and clinical data; specific the leading cause of AGE in the United States; other laboratory testing protocols vary by health depart- viral causes include adenovirus (specifically group F ment. Outbreak etiologies are considered confirmed or types 40 and 41), astrovirus, sapovirus, and rotavi- when >2 laboratory-confirmed cases are reported rus (2,3). These viruses are spread primarily through and considered suspected when <2 laboratory-con- the fecal–oral route through person-to-person contact firmed cases are reported. Outbreaks are considered or through contaminated food, water, or fomites (4–8). -

Package Insert Astrovirus, Sapovirus Open System PCR Reagents 450-052-Series
Package Insert Astrovirus, Sapovirus Open System PCR Reagents 450-052-Series For Research Use Only Research use only reagents are not intended for human or animal diagnostic use. It is the responsibility of the end user to determine the performance of the reagents in an appropriately designed validation study for their intended use. The Astrovirus, Sapovirus real-time PCR-based detection reagent is manufactured and packaged as an open system reagent (OSR) for use with open system platforms and has to be validated by the user. Examples of open system platforms are the Applied Biosystems QuantStudioTM 5 (Design & Analysis software version 1.5.1 or later), Applied Biosystems 7500 Fast Dx (SDS software version 1.4 or later), Bio-Rad CFX96 TouchTM or CFX384 TouchTM (Maestro software version 1.1 or later) real-time PCR platforms. PLEASE READ ENTIRE PACKAGE INSERT BEFORE PROCEEDING TO USE THE OSR. PRODUCT OVERVIEW The BioGX Sample-Ready™ OSR has been formulated in lyophilized format for the multiplex real-time PCR-based detection of RNA from Astrovirus (ORF1a gene1), Sapovirus (polymerase/capsid junction gene1) and a synthetic single stranded RNA [Internal Amplification Control (IAC)/Sample Processing Control (SPC)]. The synthetic single stranded RNA serves as both a sample processing control and an internal amplification control. Two different formats for the lyophilized Sample-Ready OSR kits are available: 1. BD MAXTM System REF 450-052-C-MAX 2. ABI QuantStudioTM 5, ABI 7500 Fast Dx, Bio-Rad CFX96 TouchTM and Bio-Rad CFX384 TouchTMPlatforms REF 450-052-LMP Note: BD MAXTM System OSR (450-052-C-MAX) contains all PCR primers, probes, enzymes, dNTPs, MgCl2, buffers, and other components required for PCR reaction. -

Virus World As an Evolutionary Network of Viruses and Capsidless Selfish Elements
Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements Koonin, E. V., & Dolja, V. V. (2014). Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements. Microbiology and Molecular Biology Reviews, 78(2), 278-303. doi:10.1128/MMBR.00049-13 10.1128/MMBR.00049-13 American Society for Microbiology Version of Record http://cdss.library.oregonstate.edu/sa-termsofuse Virus World as an Evolutionary Network of Viruses and Capsidless Selfish Elements Eugene V. Koonin,a Valerian V. Doljab National Center for Biotechnology Information, National Library of Medicine, Bethesda, Maryland, USAa; Department of Botany and Plant Pathology and Center for Genome Research and Biocomputing, Oregon State University, Corvallis, Oregon, USAb Downloaded from SUMMARY ..................................................................................................................................................278 INTRODUCTION ............................................................................................................................................278 PREVALENCE OF REPLICATION SYSTEM COMPONENTS COMPARED TO CAPSID PROTEINS AMONG VIRUS HALLMARK GENES.......................279 CLASSIFICATION OF VIRUSES BY REPLICATION-EXPRESSION STRATEGY: TYPICAL VIRUSES AND CAPSIDLESS FORMS ................................279 EVOLUTIONARY RELATIONSHIPS BETWEEN VIRUSES AND CAPSIDLESS VIRUS-LIKE GENETIC ELEMENTS ..............................................280 Capsidless Derivatives of Positive-Strand RNA Viruses....................................................................................................280 -

The Intestinal Virome of Malabsorption Syndrome-Affected and Unaffected
Virus Research 261 (2019) 9–20 Contents lists available at ScienceDirect Virus Research journal homepage: www.elsevier.com/locate/virusres The intestinal virome of malabsorption syndrome-affected and unaffected broilers through shotgun metagenomics T ⁎ Diane A. Limaa, , Samuel P. Cibulskib, Caroline Tochettoa, Ana Paula M. Varelaa, Fabrine Finklera, Thais F. Teixeiraa, Márcia R. Loikoa, Cristine Cervac, Dennis M. Junqueirad, Fabiana Q. Mayerc, Paulo M. Roehea a Laboratório de Virologia, Departamento de Microbiologia, Imunologia e Parasitologia, Instituto de Ciências Básicas da Saúde (ICBS), Universidade Federal do Rio Grande do Sul (UFRGS), Porto Alegre, RS, Brazil b Laboratório de Virologia, Faculdade de Veterinária, Universidade Federal do Rio Grande do Sul, Porto Alegre, RS, Brazil c Laboratório de Biologia Molecular, Instituto de Pesquisas Veterinárias Desidério Finamor (IPVDF), Eldorado do Sul, RS, Brazil d Centro Universitário Ritter dos Reis - UniRitter, Health Science Department, Porto Alegre, RS, Brazil ARTICLE INFO ABSTRACT Keywords: Malabsorption syndrome (MAS) is an economically important disease of young, commercially reared broilers, Enteric disorders characterized by growth retardation, defective feather development and diarrheic faeces. Several viruses have Virome been tentatively associated to such syndrome. Here, in order to examine potential associations between enteric Broiler chickens viruses and MAS, the faecal viromes of 70 stool samples collected from diseased (n = 35) and healthy (n = 35) High-throughput sequencing chickens from seven flocks were characterized and compared. Following high-throughput sequencing, a total of 8,347,319 paired end reads, with an average of 231 nt, were generated. Through analysis of de novo assembled contigs, 144 contigs > 1000 nt were identified with hits to eukaryotic viral sequences, as determined by GenBank database. -

Chronic Viral Infections Vs. Our Immune System: Revisiting Our View of Viruses As Pathogens
Chronic Viral Infections vs. Our Immune System: Revisiting our view of viruses as pathogens Tiffany A. Reese Assistant Professor Departments of Immunology and Microbiology Challenge your idea of classic viral infection and disease • Define the microbiome and the virome • Brief background on persistent viruses • Illustrate how viruses change disease susceptibility – mutualistic symbiosis – gene + virus = disease phenotype – virome in immune responses Bacteria-centric view of the microbiome The microbiome defined Definition of microbiome – Merriam-Webster 1 :a community of microorganisms (such as bacteria, fungi, and viruses) that inhabit a particular environment and especially the collection of microorganisms living in or on the human body 2 :the collective genomes of microorganisms inhabiting a particular environment and especially the human body Virome Ø Viral component of the microbiome Ø Includes both commensal and pathogenic viruses Ø Viruses that infect host cells Ø Virus-derived elements in host chromosomes Ø Viruses that infect other organisms in the body e.g. phage/bacteria Viruses are everywhere! • “intracellular parasites with nucleic acids that are capable of directing their own replication and are not cells” – Roossinck, Nature Reviews Microbiology 2011. • Viruses infect all living things. • We are constantly eating and breathing viruses from our environment • Only a small subset of viruses cause disease. • We even carry viral genomes as part of our own genetic material! Diverse viruses all over the body Adenoviridae Picornaviridae -

Discovery of Novel Anelloviruses in Small Mammals Expands the Host
Virology 514 (2018) 9–17 Contents lists available at ScienceDirect Virology journal homepage: www.elsevier.com/locate/virology Discovery of novel anelloviruses in small mammals expands the host range and diversity of the Anelloviridae T ⁎ William Marciel de Souzaa,b, ,1, Marcílio Jorge Fumagallia,1, Jansen de Araujoc, Gilberto Sabino-Santos Jr.a, Felipe Gonçalves Motta Maiaa,d, Marilia Farignoli Romeiroa, Sejal Modhab, Marcello Schiavo Nardie, Luzia Helena Queirozf, Edison Luiz Durigonc, Márcio Roberto Teixeira Nunesg,h, Pablo Ramiro Murciab, Luiz Tadeu Moraes Figueiredoa a Virology Research Center, Ribeirão Preto Medical School, University of São Paulo, Ribeirão Preto, Brazil b MRC-University of Glasgow Centre for Virus Research, Glasgow, United Kingdom c Laboratory Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brazil d Department of Microbiology, Institute of Biomedical Sciences, University of São Paulo, São Paulo, Brazil e Divisão Técnica de Medicina Veterinária e Manejo da Fauna Silvestre, Prefeitura de São Paulo, Brazil f Faculty of Veterinary Medicine, São Paulo State University, Araçatuba, Brazil g Center for Technological Innovations, Evandro Chagas Institute, Ministry of Health, Ananindeua, Pará, Brazil h Department of Pathology, University of Texas Medical Branch, Galveston, TX, USA ARTICLE INFO ABSTRACT Keywords: The Anelloviridae comprises single-stranded DNA viruses currently grouped in sixty-eight species classified in Anellovirus twelve genera. They have been found in many vertebrate hosts including primates. In this study, we describe the Rodent-borne virus application of the high-throughput sequencing to examine the frequency and diversity of anelloviruses in ro- Bat-borne virus dents, bats and opossums captured in São Paulo State, Brazil. -
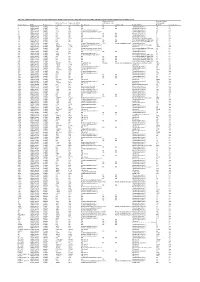
Table S4. Comparison Between the Discvr Results and the BLAST
Table S4. Comparison between the DisCVR results and the BLAST results from the original manuscript for healthy patients and patients with unexplained acute febrile illness Number of k-mers Number of reads Number of k-mers Number of distinct matching the 2nd matching with Sample_Name RUN Patient status matching k-mers matching Top Virus hits hit 2nd Virus Hit Top BLAST hit BLAST Concordance 3 SRR1748140 healthy NA NA NA NA NA Human.adenovirus.C 10 no 4 SRR1748141 healthy NA NA NA NA NA Human.adenovirus.C 65 no 8 SRR1748143 healthy 943 379 Human mastadenovirus C Human.adenovirus.C 65 yes 10 SRR1748147 healthy 1669 133 Human immunodeficiency virus 1 Human.adenovirus.C 26 no 14 SRR1748151 healthy NA NA NA NA NA Human.adenovirus.C 4 no 15 SRR1748152 healthy NA NA NA NA NA Human.adenovirus.C 6 no 108 SRR1748162 healthy NA NA NA NA NA Human.adenovirus.C 6 no 137 SRR1748172 healthy 15343 4714 Human immunodeficiency virus 1 Human.immunodeficiency.virus 314 yes 173 SRR1748173 healthy NA NA NA NA NA Heterosigma.akashiwo.RNA.virus 20 no 245 SRR1748177 healthy NA NA NA NA NA Heterosigma.akashiwo.RNA.virus 17 no 312 SRR1748178 healthy 2843 4 Human T-lymphotropic virus 1 1282 Human mastadenovirus C Human.adenovirus.C 70 no 316 SRR1748179 healthy 22915 5941 Human immunodeficiency virus 1 Human.immunodeficiency.virus 342 yes 319 SRR1748180 healthy 2247683 21201 GB virus C GB.virus.C 20629 yes 325 SRR1748181 healthy 1006 177 Human immunodeficiency virus 1 Heterosigma.akashiwo.RNA.virus 60 no 329 SRR1748182 healthy 1513 124 Human immunodeficiency virus 1 -

Detection and Characterization of a Novel Norovirus in Bats, China
Virologica Sinica (2018) 33:100–103 www.virosin.org https://doi.org/10.1007/s12250-018-0010-9 www.springer.com/12250 (0123456789().,-volV)(0123456789().,-volV) LETTER Detection and Characterization of a Novel Norovirus in Bats, China Ling’en Yang1,2 · Quanxi Wang1 · Lin Xu2 · Changchun Tu1,2,3 · Xiaohong Huang1 · Biao He2,3 Received: 7 November 2017 / Accepted: 21 December 2017 © Wuhan Institute of Virology, CAS and Springer Nature Singapore Pte Ltd. 2018. This article is an open access publication Dear Editor, found in GI, GII and GIV, while porcine NoVs group in distinct genotypes within GII, bovine and ovine viruses Noroviruses (NoVs) are second only to the rotaviruses as belong exclusively to GIII, murine NoVs are grouped in etiologic agents of acute fulminant gastroenteritis in infants GV, canine NoVs are in GIV and GVI, and lion viruses and young children worldwide, with an estimated 200,000 have been found only in GIV (Green 2013). deaths per year in children younger than 5 years of age in As one of the most widely distributed mammals, bats are developing countries (Patel et al. 2008). NoVs are classi- important natural reservoirs of viruses, of which more than fied within the genus Norovirus of the family Caliciviridae 137 have been discovered (Luis et al. 2013), including with Norwalk virus as its prototype member (ICTV 2017). many highly pathogenic agents such as Hendra and Nipah The virions are small (38–40 nm in diameter) nonen- viruses (Yob et al. 2001), SARS-related coronaviruses (Ge veloped, with an icosahedral capsidanda linear, positive- et al.