Organic Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Elimination Reactions E1, E2, E1cb and Ei
Elimination Reactions Dr. H. Ghosh Surendranath College, Kol-9 ________________________________________________ E1, E2, E1cB and Ei (pyrolytic syn eliminations); formation of alkenes and alkynes; mechanisms (with evidence), reactivity, regioselectivity (Saytzeff/Hofmann) and stereoselectivity; comparison between substitution and elimination. Substitution Reactions Elimination Reactions Elimination happens when the nucleophile attacks hydrogen instead of carbon Strong Base favor Elimination Bulky Nucleophile/Base favor Elimination High Temperature favors Elimination We know- This equation says that a reaction in which ΔS is positive is more thermodynamically favorable at higher temperature. Eliminations should therefore be favoured at high temperature Keep in Mind---- Mechanism Classification E1 Mechanism- Elimination Unimolecular E1 describes an elimination reaction (E) in which the rate-determining step is unimolecular (1) and does not involve the base. The leaving group leaves in this step, and the proton is removed in a separate second step E2 Mechanism- Elimination Bimolecular E2 describes an elimination (E) that has a bimolecular (2) rate-determining step that must involve the base. Loss of the leaving group is simultaneous with removal of the proton by the base Bulky t-butoxide—ideal for promoting E2 as it’s both bulky and a strong base (pKaH = 18). Other Organic Base used in Elimination Reaction These two bases are amidines—delocalization of one nitrogen’s lone pair on to the other, and the resulting stabilization of the protonated amidinium ion, E1 can occur only with substrates that can ionize to give relatively stable carbocations—tertiary, allylic or benzylic alkyl halides, for example. E1-Elimination Reaction not possible here The role of the leaving group Since the leaving group is involved in the rate-determining step of both E1 and E2, in general, any good leaving group will lead to a fast elimination. -

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Brittain-DR-1965-Phd-Thesis.Pdf
POLYMETHYLENE PYRIDINES , A thesis submitted by David Robert Brittain in partial fulfilment of the requirements for the degree of DOCTOR OP PHILOSOPHY in the University of London Organic Chemistry Department, dune, 1965. Imperial College, LONDON, S.W.7. ABSTRACT This thesis describes a series of attempts to syn- thesise 2,5- and 1,4-polymethylene bridged pyridines. Nuclear magnetic resonance theory predicts that protons, which are held directly over an aromatic ring, will be abnormally shielded compared with protons in aliphatic straight-chain hydrocarbons. This prediction has been verified for the central methylene protons of paracyclo- phanes. The degree of shielding, expressed in terms of the distance from the aromatic ring, is a measure of the induced ring current and hence the aromaticity of the benzene ring. Similar measurements upon 2,5- or 1,4— polymethylene bridged pyridines would make it possible to determine the degree of aromaticity of the pyridine ring relative to benzene. A review of the subject of aromaticity is presented in which special reference has been made to its inter- pretation by nuclear magnetic resonance. The synthetic work has not been brougL.t to a truly satisfactory conclusion. However, the synthetic routes to 2,5-dialkylpyridines have been thoroughly investigated and a wide variety of such compounds prepared. The functional groups at the ends of the alkyl chains have been varied in an effort to produce a derivative which would cyclise to give a 2,5-bridged pyridine. The attempted intramolecular oxidative coupling of 2,5-dihex- 51 -ynylpyridine received much attention. In the attempts to obtain a 1,4-bridged pyridine, two tricyclic compounds, each containing two quaternised pyridine rings linked by polymethylene chains, were obtained. -

Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1
Chapter 7, Haloalkanes, Properties and Substitution Reactions of Haloalkanes Table of Contents 1. Alkyl Halides (Haloalkane) 2. Nucleophilic Substitution Reactions (SNX, X=1 or 2) 3. Nucleophiles (Acid-Base Chemistry, pka) 4. Leaving Groups (Acid-Base Chemistry, pka) 5. Kinetics of a Nucleophilic Substitution Reaction: An SN2 Reaction 6. A Mechanism for the SN2 Reaction 7. The Stereochemistry of SN2 Reactions 8. A Mechanism for the SN1 Reaction 9. Carbocations, SN1, E1 10. Stereochemistry of SN1 Reactions 11. Factors Affecting the Rates of SN1 and SN2 Reactions 12. --Eliminations, E1 and E2 13. E2 and E1 mechanisms and product predictions In this chapter we will consider: What groups can be replaced (i.e., substituted) or eliminated The various mechanisms by which such processes occur The conditions that can promote such reactions Alkyl Halides (Haloalkane) An alkyl halide has a halogen atom bonded to an sp3-hybridized (tetrahedral) carbon atom The carbon–chlorine and carbon– bromine bonds are polarized because the halogen is more electronegative than carbon The carbon-iodine bond do not have a per- manent dipole, the bond is easily polarizable Iodine is a good leaving group due to its polarizability, i.e. its ability to stabilize a charge due to its large atomic size Generally, a carbon-halogen bond is polar with a partial positive () charge on the carbon and partial negative () charge on the halogen C X X = Cl, Br, I Different Types of Organic Halides Alkyl halides (haloalkanes) sp3-hybridized Attached to Attached to Attached -

Aromaticity Sem- Ii
AROMATICITY SEM- II In 1931, German chemist and physicist Sir Erich Hückel proposed a theory to help determine if a planar ring molecule would have aromatic properties .This is a very popular and useful rule to identify aromaticity in monocyclic conjugated compound. According to which a planar monocyclic conjugated system having ( 4n +2) delocalised (where, n = 0, 1, 2, .....) electrons are known as aromatic compound . For example: Benzene, Naphthalene, Furan, Pyrrole etc. Criteria for Aromaticity 1) The molecule is cyclic (a ring of atoms) 2) The molecule is planar (all atoms in the molecule lie in the same plane) 3) The molecule is fully conjugated (p orbitals at every atom in the ring) 4) The molecule has 4n+2 π electrons (n=0 or any positive integer Why 4n+2π Electrons? According to Hückel's Molecular Orbital Theory, a compound is particularly stable if all of its bonding molecular orbitals are filled with paired electrons. - This is true of aromatic compounds, meaning they are quite stable. - With aromatic compounds, 2 electrons fill the lowest energy molecular orbital, and 4 electrons fill each subsequent energy level (the number of subsequent energy levels is denoted by n), leaving all bonding orbitals filled and no anti-bonding orbitals occupied. This gives a total of 4n+2π electrons. - As for example: Benzene has 6π electrons. Its first 2π electrons fill the lowest energy orbital, and it has 4π electrons remaining. These 4 fill in the orbitals of the succeeding energy level. The criteria for Antiaromaticity are as follows: 1) The molecule must be cyclic and completely conjugated 2) The molecule must be planar. -

Classifying Halogenoalkanes Reactions Of
10 Halogenoalkanes H Naming Halogenoalkanes H H H H C H H H H Based on original alkane, with a prefix indicating halogen atom: H C C C H Fluoro for F; Chloro for Cl; Bromo for Br; Iodo for I. H C C C C H Br H H H Cl H H Substituents are listed alphabetically 1-bromopropane 2-chloro-2-methylbutane Classifying Halogenoalkanes Halogenoalkanes can be classified as primary, secondary or tertiary depending on the number of carbon atoms attached to the C-X functional group. H H H H H H H H C H H H H H C C C H H C C C H H C C C C H Br H H H Br H H Cl H H Primary halogenoalkane Secondary halogenoalkane Tertiary halogenoalkane One carbon attached to the Two carbons attached to the Three carbons attached to the carbon atom adjoining the carbon atom adjoining the carbon atom adjoining the halogen halogen halogen Reactions of Halogenoalkanes Halogenoalkanes undergo either substitution or elimination reactions Nucleophilic substitution reactions Substitution: swapping a halogen atom for another atom or groups of atoms - - Nucleophile: electron pair donator e.g. :OH , :NH3, CN :Nu represents any nucleophile – they The Mechanism: We draw (or outline) mechanisms to always have a lone pair and act as show in detail how a reaction proceeds electron pair donators Nu:- The nucleophiles The carbon has a small attack the positive H H H H positive charge because carbon atom of the electronegativity H C C X H C C + X- δ+ δ- Nu difference between the carbon and the halogen H H H H We use curly arrows in mechanisms (with A curly arrow will always two line heads) to show the movement of start from a of electrons or two electrons lone pair the centre of a bond The rate of these substitution reactions depends on the strength Bond enthalpy / of the C-X bond kJmol-1 The weaker the bond, the easier it is to break and the faster the reaction. -
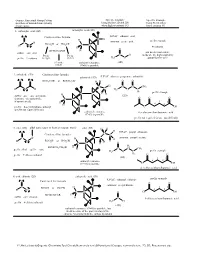
Generic Functional Group Pattern in Ordero of Nomenclature Priority in Our Course. Specific Example Using Suffix (2D and 3D)
Generic Functional Group Pattern Specific Example Specific Example in ordero of nomenclature priority Using Suffix (2D and 3D) Using Prefix when in our course. when highest priority FG lower priority FG 1. carboxylic acid (2D) carboxylic acid (3D) O Condensed line formula IUPAC: ethanoic acid O H H prefix example C H common: acetic acid RCO2H or HO2CR C C R O #-carboxy H O RCH(CO H)R' H suffix: -oic acid 2 O not used in our course FG on FG on C H (acids are the highest priority prefix: #-carboxy the right the left H3C O group that we use) FG in the carbonyl resonance (2D) middle (C=O) is possible 2. anhydride (2D) Condensed line formula anhydride (3D) IUPAC: ethanoic propanoic anhydride O O RCO2COR' or ROCO2CR' H H O O H C C C CH C 3 R O R H3C O C H H 2 prefix example H O C H suffix: -oic -oic anhydride (2D) (just one -oic anhydride, O O O C C O if symmetrical) 3 1 H 4 H 2 prefix: #-acyloxyalkanecarbonyl O O OH (prefix not required for us) carbonyl resonance 4-acyloxymethanebutanoic acid (C=O) is possible (prefix not required for us - too difficult) 3. ester (2D) alkyl name (goes in front as separate word) ester (3D) H H H IUPAC: propyl ethanoate O C Condensed line formula C O C common: propyl acetate C R' H H R O RCO2R' or R'O2CR H H C C H O H H2 RCH(CO2CH3)R' prefix: alkyl suffix: -oate H C C CH3 O H C O C prefix example 3 H prefix: #-alkoxycarbonyl 2 O O (2D) 3 1 carbonyl resonance 4 2 (C=O) is possible O OH 4-methoxycarbonylbutanoic acid 4. -

Recent Studies on the Aromaticity and Antiaromaticity of Planar Cyclooctatetraene
Symmetry 2010 , 2, 76-97; doi:10.3390/sym2010076 OPEN ACCESS symmetry ISSN 2073-8994 www.mdpi.com/journal/symmetry Review Recent Studies on the Aromaticity and Antiaromaticity of Planar Cyclooctatetraene Tohru Nishinaga *, Takeshi Ohmae and Masahiko Iyoda Department of Chemistry, Graduate School of Science and Engineering, Tokyo Metropolitan University, Hachioji, Tokyo 192-0397, Japan; E-Mails: [email protected] (T.O.); [email protected] (M.I.) * Author to whom correspondence should be addressed; E-Mail: [email protected]. Received: 29 December 2009; in revised form: 23 January 2010 / Accepted: 4 February 2010 / Published: 5 February 2010 Abstract: Cyclooctatetraene (COT), the first 4n π-electron system to be studied, adopts an inherently nonplanar tub-shaped geometry of D2d symmetry with alternating single and double bonds, and hence behaves as a nonaromatic polyene rather than an antiaromatic compound. Recently, however, considerable 8 π-antiaromatic paratropicity has been shown to be generated in planar COT rings even with the bond alternated D4h structure. In this review, we highlight recent theoretical and experimental studies on the antiaromaticity of hypothetical and actual planar COT. In addition, theoretically predicted triplet aromaticity and stacked aromaticity of planar COT are also briefly described. Keywords: antiaromaticity; cyclooctatetraene; NMR chemical shifts; quantum chemical calculations; ring current 1. Introduction Cyclooctatetraene (COT) was first prepared by Willstätter in 1911 [1,2]. At that time, the special stability of benzene was elusive and it was of interest to learn the reactivity of COT as the next higher vinylogue of benzene. However, unlike benzene, COT was found to be highly reactive to electrophiles just like other alkenes. -

Benzyl Ligand† Cite This: Chem
ChemComm View Article Online COMMUNICATION View Journal | View Issue Homoleptic organolanthanide compounds supported by the bis(dimethylsilyl)benzyl ligand† Cite this: Chem. Commun., 2017, 53,716 Kasuni C. Boteju, Arkady Ellern and Aaron D. Sadow* Received 21st November 2016, Accepted 12th December 2016 DOI: 10.1039/c6cc09304c www.rsc.org/chemcomm À A b-SiH functionalized benzyl anion [C(SiHMe2)2Ph] is obtained by A strategy for stabilizing coordinatively unsaturated rare earth deprotonation of HC(SiHMe2)2Ph with KCH2Ph or by reaction of amides has involved the incorporation of SiH groups, which 14 KOtBu and (Me2HSi)3CPh; LnI3(THF)n and three equivalents of this form labile secondary interactions with the lanthanide center. carbanion combine to provide homoleptic tris(alkyl)lanthanide Furthermore, the SiH moiety provides a powerful signature in 1 29 Creative Commons Attribution 3.0 Unported Licence. compounds Ln{C(SiHMe2)2Ph}3 (Ln = La, Ce, Pr, Nd) containing Hand Si NMR and IR spectra. This b-SiH strategy may also be secondary metal–ligand interactions. applied to alkyls, and the ligand C(SiHMe2)3 supports trivalent yttrium and divalent ytterbium and samarium homoleptic alkyls Synthesis of homoleptic organolanthanide complexes, particu- containing secondary Ln(H–Si interactions.15,16 Recently, we larly those of the early trivalent lanthanides (La–Nd), is challenging reported Ce{C(SiHMe2)3}3 as a precursor to a zwitterionic hydro- due to the large radii of these elements, polar bonding, high silylation catalyst.17 New chemistry might be accessed with alkyl charge, and high Lewis acidity.1 Such homoleptic compounds ligand variations that include both b-SiH and benzylic function- should be valuable for the synthesis of new catalysts and alities, and these groups could compete to enhance the homo- new materials,2 yet solvent- or donor-group-free, salt-free, and leptic compounds’ resistance to undesired ligand elimination This article is licensed under a thermally robust organolanthanide compounds are not readily pathways. -

Ganic Compounds
6-1 SECTION 6 NOMENCLATURE AND STRUCTURE OF ORGANIC COMPOUNDS Many organic compounds have common names which have arisen historically, or have been given to them when the compound has been isolated from a natural product or first synthesised. As there are so many organic compounds chemists have developed rules for naming a compound systematically, so that it structure can be deduced from its name. This section introduces this systematic nomenclature, and the ways the structure of organic compounds can be depicted more simply than by full Lewis structures. The language is based on Latin, Greek and German in addition to English, so a classical education is beneficial for chemists! Greek and Latin prefixes play an important role in nomenclature: Greek Latin ½ hemi semi 1 mono uni 1½ sesqui 2 di bi 3 tri ter 4 tetra quadri 5 penta quinque 6 hexa sexi 7 hepta septi 8 octa octo 9 ennea nona 10 deca deci Organic compounds: Compounds containing the element carbon [e.g. methane, butanol]. (CO, CO2 and carbonates are classified as inorganic.) See page 1-4. Special characteristics of many organic compounds are chains or rings of carbon atoms bonded together, which provides the basis for naming, and the presence of many carbon- hydrogen bonds. The valency of carbon in organic compounds is 4. Hydrocarbons: Compounds containing only the elements C and H. Straight chain hydrocarbons are named according to the number of carbon atoms: CH4, methane; C2H6 or H3C-CH3, ethane; C3H8 or H3C-CH2-CH3, propane; C4H10 or H3C-CH2- CH2-CH3, butane; C5H12 or CH3CH2CH2CH2CH3, pentane; C6H14 or CH3(CH2)4CH3, hexane; C7H16, heptane; C8H18, octane; C9H20, nonane; C10H22, CH3(CH2)8CH3, decane. -

Mann Et Al. Text 22
Catalytic Z-Selective Cross-Metathesis with Secondary Silyl- and Benzyl-Protected Allylic Ethers: Mechanistic Aspects and Applications to Natural Product Synthesis The MIT Faculty has made this article openly available. Please share how this access benefits you. Your story matters. Citation Mann, Tyler J., Alexander W. H. Speed, Richard R. Schrock, and Amir H. Hoveyda. “Catalytic Z-Selective Cross-Metathesis with Secondary Silyl- and Benzyl-Protected Allylic Ethers: Mechanistic Aspects and Applications to Natural Product Synthesis.” Angewandte Chemie International Edition 52, no. 32 (August 5, 2013): 8395-8400. As Published http://dx.doi.org/10.1002/anie.201302538 Publisher Wiley Blackwell Version Original manuscript Citable link http://hdl.handle.net/1721.1/84085 Terms of Use Creative Commons Attribution-Noncommercial-Share Alike 3.0 Detailed Terms http://creativecommons.org/licenses/by-nc-sa/3.0/ Catalytic Z-Selective Cross-Metathesis with Secondary Silyl- and Benzyl-Protected Allylic Ethers: Mechanistic Aspects and Applications to Natural Product Synthesis** Tyler J. Mann, Alexander W. H. Speed, Richard R. Schrock and Amir H. Hoveyda* [*] Prof. A. H. Hoveyda, T. J. Mann, Dr. A. W. H. Speed Department of Chemistry, Merkert Chemistry Center, Boston College, Chestnut Hill, MA 02467 (USA) Fax: (1) 617-552-1442 E-mail: [email protected] Prof. R. R. Schrock Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139 (USA) [**] Financial support was provided by the NIH (GM-59426). We are grateful to Robert V. O’Brien -

Electrochemistry and Photoredox Catalysis: a Comparative Evaluation in Organic Synthesis
molecules Review Electrochemistry and Photoredox Catalysis: A Comparative Evaluation in Organic Synthesis Rik H. Verschueren and Wim M. De Borggraeve * Department of Chemistry, Molecular Design and Synthesis, KU Leuven, Celestijnenlaan 200F, box 2404, 3001 Leuven, Belgium; [email protected] * Correspondence: [email protected]; Tel.: +32-16-32-7693 Received: 30 March 2019; Accepted: 23 May 2019; Published: 5 June 2019 Abstract: This review provides an overview of synthetic transformations that have been performed by both electro- and photoredox catalysis. Both toolboxes are evaluated and compared in their ability to enable said transformations. Analogies and distinctions are formulated to obtain a better understanding in both research areas. This knowledge can be used to conceptualize new methodological strategies for either of both approaches starting from the other. It was attempted to extract key components that can be used as guidelines to refine, complement and innovate these two disciplines of organic synthesis. Keywords: electrosynthesis; electrocatalysis; photocatalysis; photochemistry; electron transfer; redox catalysis; radical chemistry; organic synthesis; green chemistry 1. Introduction Both electrochemistry as well as photoredox catalysis have gone through a recent renaissance, bringing forth a whole range of both improved and new transformations previously thought impossible. In their growth, inspiration was found in older established radical chemistry, as well as from cross-pollination between the two toolboxes. In scientific discussion, photoredox catalysis and electrochemistry are often mentioned alongside each other. Nonetheless, no review has attempted a comparative evaluation of both fields in organic synthesis. Both research areas use electrons as reagents to generate open-shell radical intermediates. Because of the similar modes of action, many transformations have been translated from electrochemical to photoredox methodology and vice versa.