Clinical Vignette Abstracts NASPGHAN 2017
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Case Study of Distinctive Phenotypes Arising from Emanuel Syndrome in Two Karyotypically Identical Patients Mot Yee Yik1, Rabiatul Basria S.M.N
Malaysian Journal of Medicine and Health Sciences (eISSN 2636-9346) CASE REPORT A Case Study of Distinctive Phenotypes Arising From Emanuel Syndrome in Two Karyotypically Identical Patients Mot Yee Yik1, Rabiatul Basria S.M.N. Mydin2, Emmanuel Jairaj Moses1, Shahrul Hafiz Mohd Zaini2,3, Abdul Rahman Azhari4, Narazah Mohd Yusoff1 1 Regenerative Medicine Sciences Cluster, Advanced Medical and Dental Institute, Universiti Sains Malaysia, 13200 Bertam, Kepala Batas, Pulau Pinang Malaysia 2 Oncological and Radiological Sciences Cluster, Advanced Medical and Dental Institute, Universiti Sains Malaysia, 13200 Bertam, Kepala Batas, Pulau Pinang Malaysia, 3 Institute of Biological Sciences, Faculty of Science, University of Malaya, 50603 Kuala Lumpur. 4 Human Genetics Unit, Advanced Diagnostic Laboratory, Advanced Medical and Dental Institute, Universiti Sains Malaysia ABSTRACT Emanuel syndrome, also referred to as supernumerary der(22) or t(11;22) syndrome, is a rare genomic syndrome. Patients are normally presented with multiple congenital anomalies and severe developmental disabilities. Affected newborns usually carry a derivative chromosome 22 inherited from either parent, which stems from a balanced translocation between chromosomes 11 and 22. Unfortunately, identification of Emanuel syndrome carriers is diffi- cult as balanced translocations do not typically present symptoms. We identified two patients diagnosed as Emanuel syndrome with identical chromosomal aberration: 47,XX,+der(22)t(11;22)(q24;q12.1)mat karyotype but presenting variable phenotypic features. Emanuel syndrome patients present variable phenotypes and karyotypes have also been inconsistent albeit the existence of a derivative chromosome 22. Our data suggests that there may exist ac- companying genetic aberrations which influence the outcome of Emanuel syndrome phenotypes but it should be cautioned that more patient observations, diagnostic data and research is required before conclusions can be drawn on definitive karyotypic-phenotypic correlations. -

Anorectal Disorders Satish S
Gastroenterology 2016;150:1430–1442 Anorectal Disorders Satish S. C. Rao,1 Adil E. Bharucha,2 Giuseppe Chiarioni,3,4 Richelle Felt-Bersma,5 Charles Knowles,6 Allison Malcolm,7 and Arnold Wald8 1Division of Gastroenterology and Hepatology, Augusta University, Augusta, Georgia; 2Department of Gastroenterology and Hepatology, Mayo College of Medicine, Rochester, Minnesota; 3Division of Gastroenterology of the University of Verona, Azienda Ospedaliera Universitaria Integrata di Verona, Verona, Italy; 4Division of Gastroenterology and Hepatology and UNC Center for Functional GI and Motility Disorders, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina; 5Department of Gastroenterology/Hepatology, VU Medical Center, Amsterdam, The Netherlands; 6National Centre for Bowel Research and Surgical Innovation, Blizard Institute, Queen Mary University of London, London, United Kingdom; 7Division of Gastroenterology, Royal North Shore Hospital, and University of Sydney, Sydney, Australia; 8Division of Gastroenterology, University of Wisconsin School of Medicine and Public Health, Madison, Wisconsin This report defines criteria and reviews the epidemiology, questionnaires and bowel diaries are correlated,5 some pathophysiology, and management of the following com- patients may not accurately recall bowel symptoms6; hence, mon anorectal disorders: fecal incontinence (FI), func- symptom diaries may be more reliable. tional anorectal pain, and functional defecation disorders. In this report, we examine the prevalence and patho- FI is defined as the recurrent uncontrolled passage of fecal physiology of anorectal disorders, listed in Table 1,and material for at least 3 months. The clinical features of FI provide recommendations for diagnostic evaluation and are useful for guiding diagnostic testing and therapy. management. These supplement practice guidelines rec- ANORECTAL Anorectal manometry and imaging are useful for evalu- ommended by the American Gastroenterological Associa- fl ating anal and pelvic oor structure and function. -

Statistical Analysis Plan
Title: Clinical effectiveness and safety of vedolizumab intravenous in real world clinical practice in ulcerative colitis Korean patients: a multicenter postmarketing observational study NCT Number: NCT03535649 SAP Approve Date: 03 DEC 2018 Certain information within this Statistical Analysis Plan has been redacted (ie, specific content is masked irreversibly from view with a black/blue bar) to protect either personally identifiable (PPD) information or company confidential information (CCI). This may include, but is not limited to, redaction of the following: Named persons or organizations associated with the study. Proprietary information, such as scales or coding systems, which are considered confidential information under prior agreements with license holder. Other information as needed to protect confidentiality of Takeda or partners, personal information, or to otherwise protect the integrity of the clinical study. CCI Statistical Analysis Plan Page 1 of 60 Statistical Analysis Plan STUDY ID: VEDOLIZUMAB-5045 TITLE: C LINICAL EFFECTIVENESS AND SAFETY OF VEDOLIZUMAB INTRAVENOUS IN REAL WORLD CLINICAL PRACTICE IN ULCERATIVE COLITIS KOREAN PATIENTS: A MULTICENTER POST-MARKETING OBSERVATIONAL STUDY SHORT TITLE: VEDOLIZUMAB IN ULCERATIVE COLITIS KOREAN PATIENTS Prepared for: Takeda Pharmaceuticals Korea Co., Ltd. PPD AUTHOR: VERSION NUMBER AND DATE: V2.0; 03 DEC 2018 Property of Takeda: For non-commercial use only and subject to the applicable Terms of Use Document: Takeda_SAP_Vedolizumab-5045_v2.0_20181203.docx Author: PPD Version -

Zebrafish Models of Neurology Biochimica Et Biophysica Acta
Biochimica et Biophysica Acta 1812 (2011) 333–334 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbadis Editorial Preface: Zebrafish Models of Neurology☆ Forming hypotheses about the Molecular Basis of Neurological benefitted primarily from biochemistry/ biophysics of protein Disease has traditionally been based on human genetics, biochemistry aggregation, cell culture work and mouse models; These paradigms and molecular biology, whereas testing these hypotheses requires us are challenging to implement in a fashion that tells us how disease in to turn to cell culture and animal models. Zebrafish have emerged as a one portion of the CNS spreads to another. Zebrafish are positioned tractable platform to complement and bridge these paradigms in brilliantly to fill this gap, not only because of its efficiency as a genetic many diseases, allowing integration of human genetics and cell and neuroscience model, but because zebrafish have a long history as biology within the in vivo setting. Perhaps it is in Neurology and a platform for assessing non-cell-autonomous effects of protein Neurodegeneration, however, where zebrafish have special promise perturbations in the in vivo setting. Thus it is common for zebrafish to make contributions. Four special advantages of zebrafish to biologists to ask if changing gene abundance or character in one group Neurology are worth highlighting here: efficient in vivo tests for of neurons is able to affect the fate and function of neighboring cells. genetic interactions, conserved cellular architecture of the CNS, the Creative and ambitious deployment of this ability will complement ability to create genetically mosaic animals, and tractable behaviour established tools to promise a deep understanding of how disease and electrophysiology to assess experimental interventions. -

Dysphagia - Pathophysiology of Swallowing Dysfunction, Symptoms, Diagnosis and Treatment
ISSN: 2572-4193 Philipsen. J Otolaryngol Rhinol 2019, 5:063 DOI: 10.23937/2572-4193.1510063 Volume 5 | Issue 3 Journal of Open Access Otolaryngology and Rhinology REVIEW ARTICLE Dysphagia - Pathophysiology of Swallowing Dysfunction, Symptoms, Diagnosis and Treatment * Bahareh Bakhshaie Philipsen Check for updates Department of Otorhinolaryngology-Head and Neck Surgery, Odense University Hospital, Denmark *Corresponding author: Dr. Bahareh Bakhshaie Philipsen, Department of Otorhinolaryngology-Head and Neck Surgery, Odense University Hospital, Sdr. Boulevard 29, 5000 Odense C, Denmark, Tel: +45 31329298, Fax: +45 66192615 the vocal folds adduct to prevent aspiration. The esoph- Abstract ageal phase is completely involuntary and consists of Difficulty swallowing is called dysphagia. There is a wide peristaltic waves [2]. range of potential causes of dysphagia. Because there are many reasons why dysphagia can occur, treatment Dysphagia is classified into the following major depends on the underlying cause. Thorough examination types: is important, and implementation of a treatment strategy should be based on evaluation by a multidisciplinary team. 1. Oropharyngeal dysphagia In this article, we will describe the mechanism of swallowing, the pathophysiology of swallowing dysfunction and different 2. Esophageal dysphagia causes of dysphagia, along with signs and symptoms asso- 3. Complex neuromuscular disorders ciated with dysphagia, diagnosis, and potential treatments. 4. Functional dysphagia Keywords Pathophysiology Dysphagia, Deglutition, Deglutition disorders, FEES, Video- fluoroscopy Swallowing is a complex process and many distur- bances in oropharyngeal and esophageal physiology including neurologic deficits, obstruction, fibrosis, struc- Introduction tural damage or congenital and developmental condi- Dysphagia is derived from the Greek phagein, means tions can result in dysphagia. Breathing difficulties can “to eat” [1]. -

Risk Factors of Biliary Peritonitis Following T-Tube Removal- the Unsolved Problem
Jemds.com Original Research Article Risk Factors of Biliary Peritonitis following T-Tube Removal- The Unsolved Problem Partha Pratim Barua1, Devid Hazarika2, Khorshid Alom Hussain3 1Associate Professor, Department of Surgery, Fakhruddin Ali Ahmed Medical College, Barpeta, Assam, India. 2Assistant Professor, Department of Surgery, Assam Medical College, Dibrugarh, Assam, India. 3Registrar, Department of Surgery, Fakhruddin Ali Ahmed Medical College, Barpeta, Assam, India. ABSTRACT BACKGROUND Gall stone disease remains one of the most common problems leading to surgical Corresponding Author: intervention. About 15% of all gall stone disease patients have stones in the common Devid Hazarika, Gunjan’s Aparna Enclave, bile duct (choledocholithiasis). Open choledocholithotomy is still widely performed, Hatigarh Chariali, Geetanagar, particularly in centres without ERCP facilities. Though primary repair of the common Guwahati-781021, Assam, India. bile duct is possible, most surgeons prefer to drain the common bile duct with T-tube. E-mail: [email protected] This avoids pressure build up in the CBD in case of oedema around the ampulla of Vater in the immediate post-operative period. Normally, the T-tube is left for 14–20 DOI: 10.14260/jemds/2019/546 days in order to allow a fibrous tract to form around it. In absences of any distal obstruction, the T-tube is removed by gentle traction in the horizontal limb. In Financial or Other Competing Interests: None. majority of the cases no complications occur after tube removal. However, in some patients, biliary peritonitis occurs with varying severity. The aim of this study is to How to Cite This Article: find out if there are yet unrecognized factors that increases the risk of biliary Barua PP, Hazarika D, Hussain KA. -

18 (2), 2015 77-82 a Case with Emanuel Syndrome
18 (2), 2015 l 77-82 DOI: 10.1515/bjmg-2015-0089 CASE REPORT A CASE WITH EMANUEL SYNDROME: EXTRA DERIVATIVE 22 CHROMOSOME INHERITED FROM THE MOTHER İkbal Atli E*, Gürkan H, Vatansever Ü, Ulusal S, Tozkir H *Corresponding Author: Emine İkbal Atli, Ph.D., Department of Medical Genetics, Faculty of Medicine, Trakya University, Campus Of Balkan, D100 Street, Edirne 22030, Turkey. Tel: +90-554-253-4030. Fax: +90- 284-223-4201. E-mail: [email protected] ABSTRACT INTRODUCTION Emanuel syndrome (ES) is a rare chromosomal Emanuel syndrome (ES) is an unbalanced disorder that is characterized by multiple congenital translocation syndrome, usually arising through a anomalies and developmental disabilities. Affected 3:1 meiosis I malsegregation during gametogenesis children are usually identified in the newborn period in a phenotypically balanced translocation normal as the offspring of balanced (11;22) translocation carrier. While the true mortality rate in Emanuel syn- carriers. Carriers of this balanced translocation usu- drome is unknown, long-term survival is possible [1]. ally have no clinical symptoms and are often identi- Emanuel syndrome is also referred to as derivative fied after the birth of offspring with an unbalanced 22 syndrome, derivative 11;22 syndrome, partial tri- form of the translocation, the supernumerary der(22) somy 11;22, or supernumerary der(22)t(11;22) syn- t(11;22) syndrome. We report a 3-year-old boy with drome [2]. In this partial duplication, 11(q23-qter) the t(11;22)(q23;q11) chromosome, transmitted in and 22(pter-q11) complex, congenital diaphragmatic an unbalanced fashion from his mother. -

Reoperative Techniques and Management in Hirschsprung Disease: a Narrative Review
14 Review Article Reoperative techniques and management in Hirschsprung disease: a narrative review Farokh R. Demehri^, Belinda H. Dickie Department of Surgery, Boston Children’s Hospital, Boston, MA, USA Contributions: (I) Conception and design: All authors; (II) Administrative support: None; (III) Provision of study materials or patients: None; (IV) Collection and assembly of data: All authors; (V) Data analysis and interpretation: All authors; (VI) Manuscript writing: All authors; (VII) Final approval of manuscript: All authors Correspondence to: Farokh R. Demehri, MD. Department of Surgery, Boston Children’s Hospital, 300 Longwood Ave, Fegan 3, Boston, MA 02115, USA. Email: [email protected]. Abstract: The majority of children who undergo operative management for Hirschsprung disease have favorable results. A subset of patients, however, have long-term dysfunctional stooling, characterized by either frequent soiling or obstructive symptoms. The evaluation and management of a child with poor function after pull-through for Hirschsprung disease should be conducted by an experienced multidisciplinary team. A systematic workup is focused on detecting pathologic and anatomic causes of pull- through dysfunction. This includes an exam under anesthesia, pathologic confirmation including a repeat biopsy, and a contrast enema, with additional studies depending on the suspected etiology. Obstructive symptoms may be due to technique-specific types of mechanical obstruction, histopathologic obstruction, or dysmotility—each of which may benefit from reoperative surgery. The causes of soiling symptoms include loss of the dentate line and damage to the anal sphincter, which generally do not benefit from revision of the pull-through, and pseudo-incontinence, which may reveal underlying obstruction. A thorough understanding of the types of complications associated with various pull-through techniques aids in the evaluation of a child with postoperative dysfunction. -

Incidental Cholecystojejunal Fistula: a Rare Complication of Gall Stone Disease
MedCrave Online Journal of Surgery Case Report Open Access Incidental cholecystojejunal fistula: a rare complication of gall stone disease Abstract Volume 8 Issue 4 - 2020 Cholecystoenteric fistula is a rare complication of gallstone disease and difficult to diagnose Vipul K Srivastava,1 Shilpi Roy,1 Ramniwas preoperatively. Among Cholecystoenteric fistula, cholecystojejunal fistulae are even rarer Meena,2 Rahul Khanna2 and only a few case reports have been published on it. Here we report a case of a 60-year 1Resident, Department of General Surgery, Institute of Medical male patient with cholecystojejunal fistula diagnosed intraoperatively while performing Sciences, India laparoscopic cholecystectomy. Fundus of the gall bladder was found to be communicating 2Professor, Department of General Surgery, Institute of Medical with proximal jejunum. We conclude that in elderly patients if the ultrasonography shows Sciences, India features of contracted gall bladder in presence of large gall stones one should consider an option of getting a computed tomography scan done preoperatively. Correspondence: Dr. Ramniwas Meena, Professor Department of General Surgery, Institute of Medical Sciences Banaras Hindu University, Varanasi–221005, UP, India, Keywords: cholecystoenteric, cholecystojejunal, fistula, gall-stones, cholecystitis Tel +919935141697, Email Received: October 25, 2020 | Published: December 17, 2020 Introduction Cholecystoenteric fistula (CEF) was first described by Courvoisier in 1890. They are a rare complication of gallstone disease and are formed due to ongoing inflammation.1 They are bilioenteric type of Internal Biliary fistula which is rare to find. Preoperative diagnosis of CEF is difficult to make with pneumobilia being the most common radiological finding.2 So here we report a rare case of cholecystojejunal fistula. -
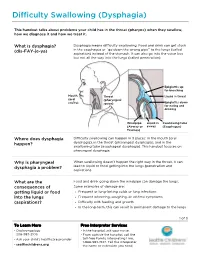
PE3334 Difficulty Swallowing (Dysphagia)
Difficulty Swallowing (Dysphagia) This handout talks about problems your child has in the throat (pharynx) when they swallow, how we diagnose it and how we treat it. What is dysphagia? Dysphagia means difficulty swallowing. Food and drink can get stuck (dis-FAY-je-ya) in the esophagus or “go down the wrong pipe” to the lungs (called aspiration) instead of the stomach. It can also go into the voice box but not all the way into the lungs (called penetration). Epiglottis up for breathing Mouth Throat Liquid in throat (oral (pharyngeal cavity) space) Epiglottis down for eating and drinking Windpipe Liquid in Swallowing tube (Airway or airway (Esophagus) Trachea) Where does dysphagia Difficulty swallowing can happen in 3 places: in the mouth (oral happen? dysphagia), in the throat (pharyngeal dysphagia), and in the swallowing tube (esophageal dysphagia). This handout focuses on pharyngeal dysphagia. Why is pharyngeal When swallowing doesn’t happen the right way in the throat, it can dysphagia a problem? lead to liquid or food getting into the lungs (penetration and aspiration). What are the Food and drink going down the windpipe can damage the lungs. consequences of Some examples of damage are: getting liquid or food • Frequent or long-lasting colds or lung infections into the lungs • Frequent wheezing, coughing, or asthma symptoms (aspiration)? • Difficulty with feeding and growth • In the long-term, this can result in permanent damage to the lungs 1 of 3 To Learn More Free Interpreter Services • Otolaryngology • In the hospital, ask your nurse. 206-987-2105 • From outside the hospital, call the • Ask your child’s healthcare provider toll-free Family Interpreting Line, 1-866-583-1527. -

The Gastrointestinal System and the Elderly
2 The Gastrointestinal System and the Elderly Thomas W. Sheehy 2.1. Introduction Gastrointestinal diseases increase with age, and their clinical presenta tions are often confused by functional complaints and by pathophysio logic changes affecting the individual organs and the nervous system of the gastrointestinal tract. Hence, the statement that diseases of the aged are characterized by chronicity, duplicity, and multiplicity is most appro priate in regard to the gastrointestinal tract. Functional bowel distress represents the most common gastrointestinal disorder in the elderly. Indeed, over one-half of all their gastrointestinal complaints are of a functional nature. In view of the many stressful situations confronting elderly patients, such as loss of loved ones, the fears of helplessness, insolvency, ill health, and retirement, it is a marvel that more do not have functional complaints, become depressed, or overcompensate with alcohol. These, of course, make the diagnosis of organic complaints all the more difficult in the geriatric patient. In this chapter, we shall deal primarily with organic diseases afflicting the gastrointestinal tract of the elderly. To do otherwise would require the creation of a sizable textbook. THOMAS W. SHEEHY • Birmingham Veterans Administration Medical Center; and University of Alabama in Birmingham, School of Medicine, Birmingham, Alabama 35233. 63 S. R. Gambert (ed.), Contemporary Geriatric Medicine © Plenum Publishing Corporation 1988 64 THOMAS W. SHEEHY 2.1.1. Pathophysiologic Changes Age leads to general and specific changes in all the organs of the gastrointestinal tract'! Invariably, the teeth show evidence of wear, dis cloration, plaque, and caries. After age 70 years the majority of the elderly are edentulous, and this may lead to nutritional problems. -

Nutrition Options in Short-Bowel Syndrome Upmcphysicianresources.Com/GI Instructions: Services
In This Issue 1 Nutrition Options in Short-Bowel Syndrome SPRING 2017 Division of Gastroenterology, 3 Gastric Carcinoids with Duodenal Ulcers Hepatology, and Nutrition 4 Living Donor Liver Transplant (LDLT) 6 PancreasFest 2017 / Honors and Awards 7 Pittsburgh Gut Club 8 What Is This? Nutrition Options in Short-Bowel Syndrome By David G. Binion, MD, and Zachary Zator, MD Intestinal transplantation is an option for select patients with short-bowel syndrome- associated intestinal failure (SBS-IF) who fail or do not tolerate nutritional rehabilitation. There are a range of factors to consider in the nutritional management of patients before and after intestinal transplantation. SBS-IF can be defined as the inability to maintain proper nutritional balance — including of proteins, electrolytes, macronutrients, micronutrients, and fluids — while adhering to a conventional diet in the face of an anatomically or functionally limited gut surface. The ideal management of patients with SBS-IF involves a multidisciplinary team of gastro enterologists, nurses, dietitians, pharmacists, and surgeons. Pharmacotherapeutic agents aimed at minimizing fluid losses have been routinely employed to support these patients. For instance, antidiarrheal agents, such as loperamide or diphenoxylate, are used alongside proton pump inhibitors. Somatostatin analogs, like octreotide, inhibit gastrointestinal secretions from the stomach, pancreas, and intestines and have been proven beneficial in the past. However, their role can be limited, as somatostatin can actually