Single Cell ADNP Predictive of Human Muscle Disorders: Mouse Knockdown Results in Muscle Wasting
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Large-Scale Opening of Utrophints Tandem Calponin Homology (CH
Large-scale opening of utrophin’s tandem calponin homology (CH) domains upon actin binding by an induced-fit mechanism Ava Y. Lin, Ewa Prochniewicz, Zachary M. James, Bengt Svensson, and David D. Thomas1 Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN 55455 Edited by James A. Spudich, Stanford University School of Medicine, Stanford, CA, and approved June 20, 2011 (received for review April 21, 2011) We have used site-directed spin labeling and pulsed electron has prevented the development of a reliable structural model for paramagnetic resonance to resolve a controversy concerning the any of these complexes. A major unresolved question concerns structure of the utrophin–actin complex, with implications for the the relative disposition of the tandem CH domains (CH1 and pathophysiology of muscular dystrophy. Utrophin is a homolog of CH2) (9, 10). Crystal structures of the tandem CH domains dystrophin, the defective protein in Duchenne and Becker muscular showed a closed conformation for fimbrin (11) and α-actinin (12), dystrophies, and therapeutic utrophin derivatives are currently but an open conformation for both utrophin (Utr261) (Fig. 1A) being developed. Both proteins have a pair of N-terminal calponin and dystrophin (Dys246) (16). The crystal structure of Utr261 homology (CH) domains that are important for actin binding. suggests that the central helical region connecting CH1 and CH2 Although there is a crystal structure of the utrophin actin-binding is highly flexible. Even for α-actinin, which has a closed crystal domain, electron microscopy of the actin-bound complexes has structure, computational analysis suggests the potential for a high produced two very different structural models, in which the CH do- degree of dynamic flexibility that facilitates actin binding (17). -

Targeted Genes and Methodology Details for Neuromuscular Genetic Panels
Targeted Genes and Methodology Details for Neuromuscular Genetic Panels Reference transcripts based on build GRCh37 (hg19) interrogated by Neuromuscular Genetic Panels Next-generation sequencing (NGS) and/or Sanger sequencing is performed Motor Neuron Disease Panel to test for the presence of a mutation in these genes. Gene GenBank Accession Number Regions of homology, high GC-rich content, and repetitive sequences may ALS2 NM_020919 not provide accurate sequence. Therefore, all reported alterations detected ANG NM_001145 by NGS are confirmed by an independent reference method based on laboratory developed criteria. However, this does not rule out the possibility CHMP2B NM_014043 of a false-negative result in these regions. ERBB4 NM_005235 Sanger sequencing is used to confirm alterations detected by NGS when FIG4 NM_014845 appropriate.(Unpublished Mayo method) FUS NM_004960 HNRNPA1 NM_031157 OPTN NM_021980 PFN1 NM_005022 SETX NM_015046 SIGMAR1 NM_005866 SOD1 NM_000454 SQSTM1 NM_003900 TARDBP NM_007375 UBQLN2 NM_013444 VAPB NM_004738 VCP NM_007126 ©2018 Mayo Foundation for Medical Education and Research Page 1 of 14 MC4091-83rev1018 Muscular Dystrophy Panel Muscular Dystrophy Panel Gene GenBank Accession Number Gene GenBank Accession Number ACTA1 NM_001100 LMNA NM_170707 ANO5 NM_213599 LPIN1 NM_145693 B3GALNT2 NM_152490 MATR3 NM_199189 B4GAT1 NM_006876 MYH2 NM_017534 BAG3 NM_004281 MYH7 NM_000257 BIN1 NM_139343 MYOT NM_006790 BVES NM_007073 NEB NM_004543 CAPN3 NM_000070 PLEC NM_000445 CAV3 NM_033337 POMGNT1 NM_017739 CAVIN1 NM_012232 POMGNT2 -
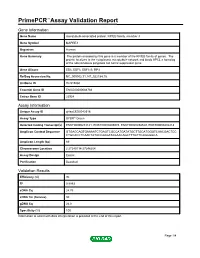
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name microtubule-associated protein, RP/EB family, member 3 Gene Symbol MAPRE3 Organism Human Gene Summary The protein encoded by this gene is a member of the RP/EB family of genes. The protein localizes to the cytoplasmic microtubule network and binds APCL a homolog of the adenomatous polyposis coli tumor suppressor gene. Gene Aliases EB3, EBF3, EBF3-S, RP3 RefSeq Accession No. NC_000002.11, NT_022184.15 UniGene ID Hs.515860 Ensembl Gene ID ENSG00000084764 Entrez Gene ID 22924 Assay Information Unique Assay ID qHsaCED0042518 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000233121, ENST00000405074, ENST00000458529, ENST00000402218 Amplicon Context Sequence GTGACCAGTGAAAATCTGAGTCGCCATGATATGCTTGCATGGGTCAACGACTCC CTGCACCTCAACTATACCAAGATAGAACAGCTTTGTTCAGGGGCA Amplicon Length (bp) 69 Chromosome Location 2:27245114-27246204 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 90 R2 0.9993 cDNA Cq 24.76 cDNA Tm (Celsius) 80 gDNA Cq 26.8 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report MAPRE3, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Identification of the Binding Partners for Hspb2 and Cryab Reveals
Brigham Young University BYU ScholarsArchive Theses and Dissertations 2013-12-12 Identification of the Binding arP tners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non- Redundant Roles for Small Heat Shock Proteins Kelsey Murphey Langston Brigham Young University - Provo Follow this and additional works at: https://scholarsarchive.byu.edu/etd Part of the Microbiology Commons BYU ScholarsArchive Citation Langston, Kelsey Murphey, "Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins" (2013). Theses and Dissertations. 3822. https://scholarsarchive.byu.edu/etd/3822 This Thesis is brought to you for free and open access by BYU ScholarsArchive. It has been accepted for inclusion in Theses and Dissertations by an authorized administrator of BYU ScholarsArchive. For more information, please contact [email protected], [email protected]. Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactions and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston A thesis submitted to the faculty of Brigham Young University in partial fulfillment of the requirements for the degree of Master of Science Julianne H. Grose, Chair William R. McCleary Brian Poole Department of Microbiology and Molecular Biology Brigham Young University December 2013 Copyright © 2013 Kelsey Langston All Rights Reserved ABSTRACT Identification of the Binding Partners for HspB2 and CryAB Reveals Myofibril and Mitochondrial Protein Interactors and Non-Redundant Roles for Small Heat Shock Proteins Kelsey Langston Department of Microbiology and Molecular Biology, BYU Master of Science Small Heat Shock Proteins (sHSP) are molecular chaperones that play protective roles in cell survival and have been shown to possess chaperone activity. -

Genetic Mutations and Mechanisms in Dilated Cardiomyopathy
Genetic mutations and mechanisms in dilated cardiomyopathy Elizabeth M. McNally, … , Jessica R. Golbus, Megan J. Puckelwartz J Clin Invest. 2013;123(1):19-26. https://doi.org/10.1172/JCI62862. Review Series Genetic mutations account for a significant percentage of cardiomyopathies, which are a leading cause of congestive heart failure. In hypertrophic cardiomyopathy (HCM), cardiac output is limited by the thickened myocardium through impaired filling and outflow. Mutations in the genes encoding the thick filament components myosin heavy chain and myosin binding protein C (MYH7 and MYBPC3) together explain 75% of inherited HCMs, leading to the observation that HCM is a disease of the sarcomere. Many mutations are “private” or rare variants, often unique to families. In contrast, dilated cardiomyopathy (DCM) is far more genetically heterogeneous, with mutations in genes encoding cytoskeletal, nucleoskeletal, mitochondrial, and calcium-handling proteins. DCM is characterized by enlarged ventricular dimensions and impaired systolic and diastolic function. Private mutations account for most DCMs, with few hotspots or recurring mutations. More than 50 single genes are linked to inherited DCM, including many genes that also link to HCM. Relatively few clinical clues guide the diagnosis of inherited DCM, but emerging evidence supports the use of genetic testing to identify those patients at risk for faster disease progression, congestive heart failure, and arrhythmia. Find the latest version: https://jci.me/62862/pdf Review series Genetic mutations and mechanisms in dilated cardiomyopathy Elizabeth M. McNally, Jessica R. Golbus, and Megan J. Puckelwartz Department of Human Genetics, University of Chicago, Chicago, Illinois, USA. Genetic mutations account for a significant percentage of cardiomyopathies, which are a leading cause of conges- tive heart failure. -

Identifying the Optimal Gene and Gene Set in Hepatocellular Carcinoma Based on Differential Expression and Differential Co-Expression Algorithm
1066 ONCOLOGY REPORTS 37: 1066-1074, 2017 Identifying the optimal gene and gene set in hepatocellular carcinoma based on differential expression and differential co-expression algorithm LI-YANG DONG1*, WEI-ZHONG ZHOU1*, JUN-WEI NI1, WEI XIANG1, WEN-HAO HU1, CHANG YU1 and HAI-YAN LI2 Departments of 1Invasive Technology and 2Rehabilitation, The First Affiliated Hospital of Wenzhou Medical University, Ouhai, Wenzhou, Zhejiang 325000, P.R. China Received June 23, 2016; Accepted August 10, 2016 DOI: 10.3892/or.2016.5333 * Abstract. The objective of this study was to identify the with ΔG = 18.681 and 24 HDE-HDC partitions in total. In optimal gene and gene set for hepatocellular carcinoma conclusion, we successfully investigated the optimal gene, (HCC) utilizing differential expression and differential MAPRE1, and gene set, nucleoside metabolic process, which co-expression (DEDC) algorithm. The DEDC algorithm may be potential biomarkers for targeted therapy and provide consisted of four parts: calculating differential expression significant insight for revealing the pathological mechanism (DE) by absolute t-value in t-statistics; computing differential underlying HCC. co-expression (DC) based on Z-test; determining optimal thresholds on the basis of Chi-squared (χ2) maximization and Introduction the corresponding gene was the optimal gene; and evaluating functional relevance of genes categorized into different Hepatocellular carcinoma (HCC) is the fifth most common partitions to determine the optimal gene set with highest mean cancer worldwide and the third leading cause of cancer-related * minimum functional information (FI) gain (ΔG). The optimal mortality (1), making it urgent to identify early diagnostic thresholds divided genes into four partitions, high DE and markers and therapeutic targets (2). -

Defining Functional Interactions During Biogenesis of Epithelial Junctions
ARTICLE Received 11 Dec 2015 | Accepted 13 Oct 2016 | Published 6 Dec 2016 | Updated 5 Jan 2017 DOI: 10.1038/ncomms13542 OPEN Defining functional interactions during biogenesis of epithelial junctions J.C. Erasmus1,*, S. Bruche1,*,w, L. Pizarro1,2,*, N. Maimari1,3,*, T. Poggioli1,w, C. Tomlinson4,J.Lees5, I. Zalivina1,w, A. Wheeler1,w, A. Alberts6, A. Russo2 & V.M.M. Braga1 In spite of extensive recent progress, a comprehensive understanding of how actin cytoskeleton remodelling supports stable junctions remains to be established. Here we design a platform that integrates actin functions with optimized phenotypic clustering and identify new cytoskeletal proteins, their functional hierarchy and pathways that modulate E-cadherin adhesion. Depletion of EEF1A, an actin bundling protein, increases E-cadherin levels at junctions without a corresponding reinforcement of cell–cell contacts. This unexpected result reflects a more dynamic and mobile junctional actin in EEF1A-depleted cells. A partner for EEF1A in cadherin contact maintenance is the formin DIAPH2, which interacts with EEF1A. In contrast, depletion of either the endocytic regulator TRIP10 or the Rho GTPase activator VAV2 reduces E-cadherin levels at junctions. TRIP10 binds to and requires VAV2 function for its junctional localization. Overall, we present new conceptual insights on junction stabilization, which integrate known and novel pathways with impact for epithelial morphogenesis, homeostasis and diseases. 1 National Heart and Lung Institute, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. 2 Computing Department, Imperial College London, London SW7 2AZ, UK. 3 Bioengineering Department, Faculty of Engineering, Imperial College London, London SW7 2AZ, UK. 4 Department of Surgery & Cancer, Faculty of Medicine, Imperial College London, London SW7 2AZ, UK. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Inhibition of Β-Catenin Signaling Respecifies Anterior-Like Endothelium Into Beating Human Cardiomyocytes Nathan J
© 2015. Published by The Company of Biologists Ltd | Development (2015) 142, 3198-3209 doi:10.1242/dev.117010 RESEARCH ARTICLE STEM CELLS AND REGENERATION Inhibition of β-catenin signaling respecifies anterior-like endothelium into beating human cardiomyocytes Nathan J. Palpant1,2,3,*, Lil Pabon1,2,3,*, Meredith Roberts2,3,4, Brandon Hadland5,6, Daniel Jones7, Christina Jones3,8,9, Randall T. Moon3,8,9, Walter L. Ruzzo7, Irwin Bernstein5,6, Ying Zheng2,3,4 and Charles E. Murry1,2,3,4,10,‡ ABSTRACT lineages. Anterior mesoderm (mid-streak) gives rise to cardiac and endocardial endothelium, whereas posterior mesoderm (posterior During vertebrate development, mesodermal fate choices are regulated streak) gives rise to the blood-forming endothelium and vasculature by interactions between morphogens such as activin/nodal, BMPs and (Murry and Keller, 2008). Wnt/β-catenin that define anterior-posterior patterning and specify Well-described anterior-posterior morphogen gradients, downstream derivatives including cardiomyocyte, endothelial and including those of activin A/nodal and BMP4, are thought to hematopoietic cells. We used human embryonic stem cells to explore pattern mesoderm subtypes (Nostro et al., 2008; Sumi et al., 2008; how these pathways control mesodermal fate choices in vitro. Varying Kattman et al., 2011). Such gradients are proposed to specify doses of activin A and BMP4 to mimic cytokine gradient polarization anterior mesodermal fates like cardiomyocytes versus posterior in the anterior-posterior axis of the embryo led to differential activity mesodermal fates like blood. Remarkably, a recent study showed of Wnt/β-catenin signaling and specified distinct anterior-like (high activin/ that ectopic induction of a nodal/BMP gradient in zebrafish embryos low BMP) and posterior-like (low activin/high BMP) mesodermal was sufficient to create an entirely new embryonic axis that could populations. -

Role of Non-Motile Microtubule-Associated Proteins in Virus Trafficking
BioMol Concepts 2016; 7(5-6): 283–292 Review Débora M. Portilho, Roger Persson and Nathalie Arhel* Role of non-motile microtubule-associated proteins in virus trafficking DOI 10.1515/bmc-2016-0018 continuously switching between phases of growth and Received June 21, 2016; accepted November 4, 2016 shrinkage, referred to as dynamic instability (1). MTs are involved in a wide range of cellular functions, including Abstract: Viruses are entirely dependent on their ability cell morphology, division, motility, and organelle posi- to infect a host cell in order to replicate. To reach their site tioning, as well as in disease, such as neurodegenerative of replication as rapidly and efficiently as possible follow- disease, infection and immunity. Once the MT filaments ing cell entry, many have evolved elaborate mechanisms are formed by nucleation at the centrosome and elon- to hijack the cellular transport machinery to propel them- gated from the subunit pool, their stability and mechani- selves across the cytoplasm. Long-range movements have cal properties are often controlled by a set of proteins that been shown to involve motor proteins along microtubules bind along the polymer. These so-called microtubule- (MTs) and direct interactions between viral proteins and associated proteins (MAPs, not to be confused with mito- dynein and/or kinesin motors have been well described. gen-activated proteins) are essential for the regulation of Although less well-characterized, it is also becoming MT dynamics. increasingly clear that non-motile microtubule-associated Several types of MAPs have been identified in eukary- proteins (MAPs), including structural MAPs of the MAP1 otes, including MT motors, MT plus-end and minus-end and MAP2 families, and microtubule plus-end tracking tracking proteins (+ TIPs, − TIPs), centrosome-associated proteins (+ TIPs), can also promote viral trafficking in proteins and structural MAPs. -

A Heterozygous Microdeletion of 20Q13.13 Encompassing ADNP Gene in a Child with Helsmoortel–Van Der Aa Syndrome
European Journal of Human Genetics (2018) 26:1497–1501 https://doi.org/10.1038/s41431-018-0165-8 ARTICLE A heterozygous microdeletion of 20q13.13 encompassing ADNP gene in a child with Helsmoortel–van der Aa syndrome 1,2 1 3 1 4 Minh-Tuan Huynh ● Elise Boudry-Labis ● Alfred Massard ● Caroline Thuillier ● Bruno Delobel ● 4 5 Bénédicte Duban-Bedu ● Catherine Vincent-Delorme Received: 8 September 2017 / Revised: 3 April 2018 / Accepted: 11 April 2018 / Published online: 13 June 2018 © European Society of Human Genetics 2018 Abstract Helsmoortel–van der Aa (SWI/SNF autism-related or ADNP syndrome) is an autosomal dominant monogenic syndrome caused by de novo variants in the last exon of ADNP gene and no deletions have been documented to date. We report the first case of a 3 years and 10 months old boy exhibiting typical features of ADNP syndrome, including intellectual disability, autistic traits, facial dysmorphism, hyperlaxity, mood disorder, behavioral problems, and severe chronic constipation. 60K Agilent array-comparative genomic hybridization (CGH) identified a heterozygous interstitial microdeletion at 20q13.13 chromosome region, encompassing ADNP and DPM1. Taking into account the clinical phenotype of previously reported cases with ADNP single-point variants, – – 1234567890();,: 1234567890();,: genotype phenotype correlation in the proband was established and the diagnosis of Helsmoortel van der Aa syndrome was made. Our report thus confirms that ADNP haploinsufficiency is associated with Helsmoortel–van der Aa syndrome as well as highlights the utility of whole-genome array-CGH for detection of unbalanced submicroscopic chromosomal rearrangements in routine clinical setting in patients with unexplained intellectual disability and/or syndromic autism. -

EB3 (MAPRE3) (NM 012326) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC204135 EB3 (MAPRE3) (NM_012326) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: EB3 (MAPRE3) (NM_012326) Human Tagged ORF Clone Tag: Myc-DDK Symbol: MAPRE3 Synonyms: EB3; EBF3; EBF3-S; RP3 Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RC204135 ORF sequence Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGCCGTCAATGTGTACTCCACATCTGTGACCAGTGAAAATCTGAGTCGCCATGATATGCTTGCATGGG TCAACGACTCCCTGCACCTCAACTATACCAAGATAGAACAGCTTTGTTCAGGGGCAGCCTACTGCCAGTT CATGGACATGCTCTTCCCCGGCTGTGTGCACTTGAGGAAAGTGAAGTTCCAGGCCAAACTAGAGCATGAA TACATCCACAACTTCAAGGTGCTGCAAGCAGCTTTCAAGAAGATGGGTGTTGACAAAATCATTCCTGTAG AGAAATTAGTGAAAGGAAAATTCCAAGATAATTTTGAGTTTATTCAGTGGTTTAAGAAATTCTTTGACGC AAACTATGATGGAAAGGATTACAACCCTCTGCTGGCGCGGCAGGGCCAGGACGTAGCGCCACCTCCTAAC CCAGGTGATCAGATCTTCAACAAATCCAAGAAACTCATTGGCACAGCAGTTCCACAGAGGACGTCCCCCA CAGGCCCAAAAAACATGCAGACCTCTGGCCGGCTGAGCAATGTGGCCCCCCCCTGCATTCTCCGGAAGAA TCCTCCATCAGCCCGAAATGGCGGCCATGAGACTGATGCCCAAATTCTTGAACTCAACCAACAGCTGGTG GACTTGAAGCTGACAGTGGATGGGCTGGAGAAGGAACGTGACTTCTACTTCAGCAAACTTCGTGACATCG AGCTCATCTGCCAGGAGCATGAAAGTGAAAACAGCCCTGTTATCTCAGGCATCATTGGCATCCTCTATGC CACAGAGGAAGGATTCGCACCCCCTGAGGACGATGAGATTGAAGAGCATCAACAAGAAGACCAGGACGAG TAC ACGCGTACGCGGCCGCTCGAGCAGAAACTCATCTCAGAAGAGGATCTGGCAGCAAATGATATCCTGGATT