The Physics of Chromatin
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Nature of Genomes Viral Genomes Prokaryotic Genome
The nature of genomes • Genomics: study of structure and function of genomes • Genome size – variable, by orders of magnitude – number of genes roughly proportional to genome size • Plasmids – symbiotic DNA molecules, not essential – mostly circular in prokaryotes • Organellar DNA – chloroplast, mitochondrion – derived by endosymbiosis from bacterial ancestors Chapter 2: Genes and genomes © 2002 by W. H. Freeman and Company Chapter 2: Genes and genomes © 2002 by W. H. Freeman and Company Viral genomes • Nonliving particle In prokaryotes, viruses are – nucleic acid sometimes referred to as – protein bacteriophages. • DNA or RNA – single-stranded or double-stranded – linear or circular • Compact genomes with little spacer DNA Chapter 2: Genes and genomes © 2002 by W. H. Freeman and Company Chapter 2: Genes and genomes © 2002 by W. H. Freeman and Company Prokaryotic genome • Usually circular double helix – occupies nucleoid region of cell – attached to plasma membrane • Genes are close together with little intergenic spacer • Operon – tandem cluster of coordinately regulated genes – transcribed as single mRNA • Introns very rare Chapter 2: Genes and genomes © 2002 by W. H. Freeman and Company Chapter 2: Genes and genomes © 2002 by W. H. Freeman and Company 1 Eukaryotic nuclear genomes • Each species has characteristic chromosome number • Genes are segments of nuclear chromosomes • Ploidy refers to number of complete sets of chromosomes –haploid (1n): one complete set of genes – diploid (2n) – polyploid (≥3n) • In diploids, chromosomes come in homologous pairs (homologs) In humans, somatic cells have – structurally similar 2n = 46 chromosomes. – same sequence of genes – may contain different alleles Chapter 2: Genes and genomes © 2002 by W. H. -

Visualization of Chromatin Folding Patterns in Chicken Erythrocytes by Atomic Force Microscopy (AFM)
Cell Research (1997), 7, 143-150 Visualization of chromatin folding patterns in chicken erythrocytes by atomic force microscopy (AFM) 1 QIAN RUO LAN ZHENG XIA LIU, MEI YUN ZHOU, HEN YUE XIE, CHU JIANG, ZHI JIANG YAN Shanghai Institute of Cell Biology, Chinese Academy of Sciences, Shanghai 200031, China LI MIN QIAN, YI ZHANG, JUN HU Shanghai Institute of Nuclear Research, Chinese Academy of Sciences, Shanghai 201800, China ABSTRACT The organization of the higher order structure of chro- matin in chicken erythrocytes has been examined with tapping-mode scanning force microscopy under conditions close to their native environment. Reproducible high- resolution AFM images of chromatin compaction at seve- ral levels can be demonstrated. An extended beads-on-a- string (width of ~ 15-20nm, height of ~ 2-3nm for each individual nucleosome) can be consistently observed. Fur- thermore, superbeads (width of ~ 40nm, height of ~ 7nm) are demonstrated. Visualization of the solenoid conforma- tion at the level of 30nm chromatin fiber is attained either by using AFM or by using electron microscopy. In addi- tion, tightly coiled chromatin fibers (~ 50-60nm and ~ 90- ll0nm) can be revealed. Our data suggest that the chro- matin in the interphase nucleus of chicken erythrocyte rep- resents a high-order conformation and AFM provides use- ful high-resolution structural information concerning the folding pattern of interphase chromatin fibers. Key words: The chromatin folding pattern, chicken erythrocyte, atomic force microscopy. 1. To whom correspondence should be addressed: Shanghai Institute of Cell Biology, Chinese Academy of Sciences, 320 Yueyang Road, Shanghai 200031, China. 143 The chromatin folding patterns in chicken erythrocytes by AFM INTRODUCTION Owing to the tremendous packing density and folding complexity in mitotic chro- mosomes, analysis of chromosome architecture has recently focused on interphase chromatin structure. -

The Origin of the Eukaryotic Cell Based on Conservation of Existing
The Origin of the Eukaryotic Albert D. G. de Roos The Beagle Armada Cell Based on Conservation Bioinformatics Division of Existing Interfaces Einsteinstraat 67 3316GG Dordrecht, The Netherlands [email protected] Abstract Current theories about the origin of the eukaryotic Keywords cell all assume that during evolution a prokaryotic cell acquired a Evolution, nucleus, eukaryotes, self-assembly, cellular membranes nucleus. Here, it is shown that a scenario in which the nucleus acquired a plasma membrane is inherently less complex because existing interfaces remain intact during evolution. Using this scenario, the evolution to the first eukaryotic cell can be modeled in three steps, based on the self-assembly of cellular membranes by lipid-protein interactions. First, the inclusion of chromosomes in a nuclear membrane is mediated by interactions between laminar proteins and lipid vesicles. Second, the formation of a primitive endoplasmic reticulum, or exomembrane, is induced by the expression of intrinsic membrane proteins. Third, a plasma membrane is formed by fusion of exomembrane vesicles on the cytoskeletal protein scaffold. All three self-assembly processes occur both in vivo and in vitro. This new model provides a gradual Darwinistic evolutionary model of the origins of the eukaryotic cell and suggests an inherent ability of an ancestral, primitive genome to induce its own inclusion in a membrane. 1 Introduction The origin of eukaryotes is one of the major challenges in evolutionary cell biology. No inter- mediates between prokaryotes and eukaryotes have been found, and the steps leading to eukaryotic endomembranes and endoskeleton are poorly understood. There are basically two competing classes of hypotheses: the endosymbiotic and the autogenic. -

Archaea and the Origin of Eukaryotes
REVIEWS Archaea and the origin of eukaryotes Laura Eme, Anja Spang, Jonathan Lombard, Courtney W. Stairs and Thijs J. G. Ettema Abstract | Woese and Fox’s 1977 paper on the discovery of the Archaea triggered a revolution in the field of evolutionary biology by showing that life was divided into not only prokaryotes and eukaryotes. Rather, they revealed that prokaryotes comprise two distinct types of organisms, the Bacteria and the Archaea. In subsequent years, molecular phylogenetic analyses indicated that eukaryotes and the Archaea represent sister groups in the tree of life. During the genomic era, it became evident that eukaryotic cells possess a mixture of archaeal and bacterial features in addition to eukaryotic-specific features. Although it has been generally accepted for some time that mitochondria descend from endosymbiotic alphaproteobacteria, the precise evolutionary relationship between eukaryotes and archaea has continued to be a subject of debate. In this Review, we outline a brief history of the changing shape of the tree of life and examine how the recent discovery of a myriad of diverse archaeal lineages has changed our understanding of the evolutionary relationships between the three domains of life and the origin of eukaryotes. Furthermore, we revisit central questions regarding the process of eukaryogenesis and discuss what can currently be inferred about the evolutionary transition from the first to the last eukaryotic common ancestor. Sister groups Two descendants that split The pioneering work by Carl Woese and colleagues In this Review, we discuss how culture- independent from the same node; the revealed that all cellular life could be divided into three genomics has transformed our understanding of descendants are each other’s major evolutionary lines (also called domains): the archaeal diversity and how this has influenced our closest relative. -

Transcription Regulation in Eukaryotes HFSP Workshop Reports
Transcription Regulation in Eukaryotes HFSP Workshop Reports Senior editor: Jennifer Altman Assistant editor: Chris Coath I. Coincidence Detection in the Nervous System, eds A. Konnerth, R. Y. Tsien, K. Mikoshiba and J. Altman (1996) II. Vision and Movement Mechanisms in the Cerebral Cortex, eds R. Caminiti, K.-P. Hoffmann, F. Laquaniti and J. Altman (1996) III. Genetic Control of Heart Development, eds R. P. Harvey, E. N. Olson, R. A. Schulz and J. S. Altman (1997) IV. Central Synapses: Quantal Mechanisms and Plasticity, eds D. S. Faber, H. Korn, S. J. Redman, S. M. Thompson and J. S. Altman (1998) V. Brain and Mind: Evolutionary Perspectives, eds M. S. Gazzaniga and J. S. Altman (1998) VI. Cell Surface Proteoglycans in Signalling and Development, eds A. Lander, H. Nakato, S. B. Selleck, J. E. Turnbull and C. Coath (1999) VII. Transcription Regulation in Eukaryotes, eds P. Chambon, T. Fukasawa, R. Kornberg and C. Coath (1999) Forthcoming VIII. Replicon Theory and Cell Division, eds M. Kohiyama, W. Fangman, T. Kishimoto and C. Coath IX. The Regulation of Sleep, eds A. A. Borbély, O. Hayaishi, T. Sejnowski and J. S. Altman X. Axis Formation in the Vertebrate Embryo, eds S. Ang, R. Behringer, H. Sasaki, J. S. Altman and C. Coath XI. Neuroenergetics: Relevance for Functional Brain Imaging, eds P. J. Magistretti, R. G. Shulman, R. S. J. Frackowiak and J. S. Altman WORKSHOP VII Transcription Regulation in Eukaryotes Copyright © 1999 by the Human Frontier Science Program Please use the following format for citations: “Transcription Regulation in Eukaryotes” Eds P. Chambon, T. Fukasawa, R. -

A 1-Dimensional Statistical Mechanics Model for Nucleosome Positioning on Genomic DNA
A 1-dimensional statistical mechanics model for nucleosome positioning on genomic DNA S. Tesoro1, I. Ali2, A. N. Morozov3, N. Sulaiman2, D. Marenduzzo3 1Theory of Condensed Matter, Cavendish Laboratory, University of Cambridge, JJ Thomson Avenue, Cambridge CB3 0HE, United Kingdom 2Department of Physics, College of Science, PO Box 36, Sultan Qaboos University, Al-Khodh 123, Oman 3SUPA, School of Physics and Astronomy, University of Edinburgh, Mayfield Road, Edinburgh EH9 3JZ E-mail: [email protected] Abstract. The first level of folding of DNA in eukaryotes is provided by the so-called \10-nm chromatin fibre”, where DNA wraps around histone proteins (∼10 nm in size) to form nucleosomes, which go on to create a zig-zagging bead-on-a-string structure. In this work we present a 1-dimensional statistical mechanics model to study nucleosome positioning within one such 10 nm fibre. We focus on the case of genomic sheep DNA, and we start from effective potentials valid at infinite dilution and determined from high-resolution in vitro salt dialysis experiments. We study positioning within a polynucleosome chain, and compare the results for genomic DNA to that obtained in the simplest case of homogeneous DNA, where the problem can be mapped to a Tonks gas [1]. First, we consider the simple, analytically solvable, case where nucleosomes are assumed to be point-like. Then, we perform numerical simulations to gauge the effect of their finite size on the nucleosomal distribution probabilities. Finally we compare nucleosome distributions and simulated nuclease digestion patterns for the two cases (homogeneous and sheep DNA), thereby providing testable predictions of the effect of sequence on experimentally observable quantities in experiments on polynucleosome chromatin fibres reconstituted in vitro. -

Abstract Flores Vergara, Miguel
ABSTRACT FLORES VERGARA, MIGUEL ANGEL. Diversity of Scaffold/Matrix Attachment Regions (S/MARs) in Arabidopsis is Revealed by Analysis of Sequence Characteristics, Nucleosome Occupancy, Epigenetic Marks, and Gene Expression. (Under the direction of Dr. George C. Allen and Dr. William F. Thompson.) Eukaryotic chromatin is organized as independent loops of varying sizes. Following histone extraction with lithium diiodosalicylate (LIS), these loops can be visualized as a DNA halo anchored to the nuclear matrix structure. As a basic unit, the loop is thought to be essential for DNA replication, transcription and chromosomal packaging. The formation of each loop is dependent on a specific chromatin segment that must function as an anchor to the nuclear matrix. Sequences that attach specifically to the nuclear matrix have been termed scaffold/matrix attachment regions (S/MARs). Since only a limited number of putative S/MARs have been characterized so far, their role in genomic structure and function is not well understood. Thus, a more global analysis is necessary to answer a variety of questions such as: How are S/MARs distributed across the genome? Are S/MARs associated with different genomic features and are S/MARs typically AT-rich, as previously suggested? What is the nucleosomal organization at S/MAR sequences and do they define regions of accessible chromatin? Are S/MARs associated with specific epigenetic features such as certain histone modifications or DNA methylation? What role do S/MARs play in transcriptional regulation? I have approached these questions by mapping the S/MARs on Arabidopsis chromosome 4 (chr4) using a high-resolution tiling array. -

Explain the Importance of Gene Regulation in Both Prokaryotes And
Unit 4 - Gene Expression and Module 4A – Control of Gene Expression Genetic Technology Every cell contains thousands of genes 4A. Control of Gene Expression which code for proteins. However, every gene is not actively 4B. Biotechnology producing proteins at all times. 4C. Genomics In this module, we will examine some 4D. Genes and Development of the factors that help regulate when a gene is active, and how strongly it is expressed. 1 2 Objective # 1 Objective 1 Prokaryotes: Explain the importance of gene ¾ are unicellular or colonial regulation in both prokaryotes ¾ evolved to quickly exploit transient and eukaryotes. resources ¾ main role of gene regulation is to allow cells to adjust to changing conditions ¾ in the same cell, different genes are active at different times 3 4 Objective 1 Objective # 2 Eukaryotes: ¾ mostly complex multicellular organisms List and describe the different ¾ evolved the ability to maintain a stable levels of gene regulation, and internal environment (homeostasis) identify the level where genes ¾ main role of gene regulation is to allow are most commonly regulated. specialization and division of labor among cells ¾ at the same time, different genes are active in different cells 5 6 1 Objective 2 Objective 2 We can classify levels of gene To be expressed, a gene must be regulation into 2 main categories: transcribed into m-RNA, the m-RNA must be translated into a protein, and ¾ Transcriptional controls - factors that the protein must become active. regulate transcription ¾ Posttranscriptional controls – factors Gene regulation can theoretically occur that regulate any step in gene at any step in this process. -
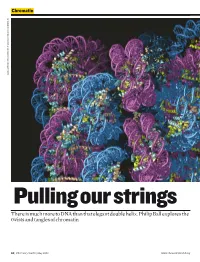
There Is Much More to DNA Than That Elegant Double Helix. Philip Ball Explores the Twists and Tangles of Chromatin
Chromatin DANIELA RHODES / MRC LABORATORY OF MOLECULAR BIOLOGY, CAMBRIDGE, UK CAMBRIDGE, BIOLOGY, MOLECULAR OF LABORATORY MRC / RHODES DANIELA Pulling our strings There is much more to DNA than that elegant double helix. Philip Ball explores the twists and tangles of chromatin 50 | Chemistry World | May 2008 www.chemistryworld.org Why isn’t my arm a leg, or my liver read and transcribed into RNA a kidney? Their cells all have the In short – the first step in the conversion same genes. But genetics isn’t just Chromosomes are of genetic information to proteins. about what genes you have – it also made up of a fibrous To understand why some genes depends on how you use them. composite of DNA and are silent while others are actively This crucial aspect of our biological protein called chromatin transcribed – and thus what allows identities has been overshadowed by This coiled network genetically identical cells to become a focus on the identity of our genes, of protein fibres plays differentiated into different tissue and how these differ from those of an important role in types – we need to unravel the other people and other species. But it controlling the activity of packaging. has now become clear that genetics an organism’s genes can’t be fully understood until we Chromatin can be Wound up genes have a better picture, not just of what changed and remodelled In eukaryotic cells, where the is in our genes, but of how they are with chemical labels DNA is housed in a cell nucleus, labelled and packaged. that provide signals for the chromosomes aren’t pure This evolving view of gene enzymes, indicating DNA. -

Chromosome Structure and Organisation
NPTEL – Biotechnology – Cell Biology Module 2- Chromosome structure and organisation This module deals with the genetic material of the cell, its structure, with details of the human chromosome and the giant chromosomes. Module 2 Lecture 1 Genetic material in a cell: All cells have the capability to give rise to new cells and the encoded information in a living cell is passed from one generation to another. The information encoding material is the genetic or hereditary material of the cell. Prokaryotic genetic material: The prokaryotic (bacterial) genetic material is usually concentrated in a specific clear region of the cytoplasm called nucleiod. The bacterial chromosome is a single, circular, double stranded DNA molecule mostly attached to the plasma membrane at one point. It does not contain any histone protein. Escherichia coli DNA is circular molecule 4.6 million base pairs in length, containing 4288 annotated protein-coding genes (organized into 2584 operons), seven ribosomal RNA (rRNA) operons, and 86 transfer RNA (tRNA) genes. Certain bacteria like the Borrelia burgdorferi possess array of linear chromosome like eukaryotes. Besides the chromosomal DNA many bacteria may also carry extra chromosomal genetic elements in the form of small, circular and closed DNA molecules, called plasmids. They generally remain floated in the cytoplasm and bear different genes based on which they have been studied. Some of the different types of plasmids are F plasmids, R plasmids, virulent plasmids, metabolic plasmids etc. Figure 1 depicts a bacterial chromosome and plasmid. Figure 1: Bacterial genetic material Joint initiative of IITs and IISc – Funded by MHRD Page 1 of 24 NPTEL – Biotechnology – Cell Biology Virus genetic material: The chromosomal material of viruses is DNA or RNA which adopts different structures. -

Review What Determines the Folding of the Chromatin Fiber?
Proc. Natl. Acad. Sci. USA Vol. 93, pp. 10548-10555, October 1996 Review What determines the folding of the chromatin fiber? Kensal van Holde*t and Jordanka Zlatanova4 *Department of Biochemistry and Biophysics, Oregon State University, Corvallis, OR 97331-7305; and PInstitute of Genetics, Bulgarian Academy of Sciences, Sofia 1113, Bulgaria ABSTRACT In this review, we attempt to summarize, in a DOES THE LINKER DNA BEND? critical manner, what is currently known about the processes of condensation and decondensation of chromatin fibers. We The solenoid model appeared to gain substantial support from begin with a critical analysis of the possible mechanisms for the studies of Yao et al. (6), which provided evidence that the condensation, considering both old and new evidence as to linker DNA in dinucleosomes did in fact contract or fold in whether the linker DNA between nucleosomes bends or re- some fashion as the salt concentration was raised from 0 to 20 mains straight in the condensed structure. Concluding that mM. The picture (and in particular, the putative role of linker the preponderance of evidence is for straight linkers, we ask histones) was complicated by a subsequent study (7), which what other fundamental process might allow condensation, showed that the same changes could be observed with dinu- and argue that there is evidence for linker histone-induced cleosomes from which linker histones had been removed. Yao contraction of the internucleosome angle, as salt concentra- et al. (6, 7) used two techniques to provide evidence for linker tion is raised toward physiological levels. We also ask how contraction: (i) direct visualization of fixed dinucleosomes by certain specific regions of chromatin can become decon- transmission electron microscopy (EM) and (ii) measurement densed, even at physiological salt concentration, to allow of the translational diffusion coefficient of dinucleosomes by transcription. -

Working with Molecular Genetics Part Four, VI=Chpt. 20. Changes in Chromatin Structure
Working with Molecular Genetics Part Four, VI=Chpt. 20. Changes in Chromatin Structure B M B 400 Part Four: Gene Regulation Section VI = Chapter 20 REGULATION BY CHANGES IN CHROMATIN STRUCTURE Review of nucleosome and chromatin structure Nucleosome composition Nucleosomes are the repeating subunit of chromatin. Nucleosomes are composed of a nucleosome core, histone H1 (in higher eukaryotes) and variable length linker DNA (0-50bp). The nucleosome core contains an octamer of 2 each of the core histones (H2A, H2B, H3 and H4) and 146 bp of DNA wrapped 1.75 turns (Fig. 4.6.1). Core histones are small basic proteins (11-14 kDa) that contain a central structure histone- fold domain and N-terminal and C-terminal extensions. Figure 4.6.1. Nucleosome core particle. A “top” view derived from the three-dimensional structure deduced in T. Richmond’s laboratory. Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. “Crystal structure of the nucleosome core particle at 2.8 A resolution.” Nature. 1997 389:251-260. Working with Molecular Genetics Part Four, VI=Chpt. 20. Changes in Chromatin Structure Histone interactions in the nucleosome Core histones dimerize through their histone fold motifs generating H3/H4 dimers and H2A H2B dimers (Fig. 4.6.2.). Two H3/H4 dimers associate to form a tetramer, which binds DNA. Two H2A/H2B dimers associate with the tetramer to form the histone octamer. At physiological salt the octamer is not stable unless bound to DNA and dissociates into the H3/H4 tetramer and two H2A/H2B dimers. Each histone pair bends approximately 30bp of DNA around the histone octamer.