Molecular Evolution and Population Genetics of Infectious Diseases
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Accurate and Rapid Identification of the Burkholderia Pseudomallei Near-Neighbour, Burkholderia Ubonensis, Using Real-Time PCR
Accurate and Rapid Identification of the Burkholderia pseudomallei Near-Neighbour, Burkholderia ubonensis, Using Real-Time PCR Erin P. Price1*, Derek S. Sarovich1, Jessica R. Webb1, Jennifer L. Ginther2, Mark Mayo1, James M. Cook2, Meagan L. Seymour2, Mirjam Kaestli1, Vanessa Theobald1, Carina M. Hall2, Joseph D. Busch2, Jeffrey T. Foster2, Paul Keim2, David M. Wagner2, Apichai Tuanyok2, Talima Pearson2, Bart J. Currie1 1 Global and Tropical Health Division, Menzies School of Health Research, Darwin, Northern Territory, Australia, 2 Center for Microbial Genetics and Genomics, Northern Arizona University, Flagstaff, Arizona, United States of America Abstract Burkholderia ubonensis is an environmental bacterium belonging to the Burkholderia cepacia complex (Bcc), a group of genetically related organisms that are associated with opportunistic but generally nonfatal infections in healthy individuals. In contrast, the near-neighbour species Burkholderia pseudomallei causes melioidosis, a disease that can be fatal in up to 95% of cases if left untreated. B. ubonensis is frequently misidentified as B. pseudomallei from soil samples using selective culturing on Ashdown’s medium, reflecting both the shared environmental niche and morphological similarities of these species. Additionally, B. ubonensis shows potential as an important biocontrol agent in B. pseudomallei-endemic regions as certain strains possess antagonistic properties towards B. pseudomallei. Current methods for characterising B. ubonensis are laborious, time-consuming and costly, and as such this bacterium remains poorly studied. The aim of our study was to develop a rapid and inexpensive real-time PCR-based assay specific for B. ubonensis. We demonstrate that a novel B. ubonensis-specific assay, Bu550, accurately differentiates B. ubonensis from B. -

Isolation, Cloning and Co-Expression of Lipase and Foldase Genes of Burkholderia Territorii GP3 from Mount Papandayan Soil
J. Microbiol. Biotechnol. (2019), 29(6), 944–951 https://doi.org/10.4014/jmb.1812.12013 Research Article Review jmb Isolation, Cloning and Co-Expression of Lipase and Foldase Genes of Burkholderia territorii GP3 from Mount Papandayan Soil Ludwinardo Putra1, Griselda Herman Natadiputri2, Anja Meryandini1, and Antonius Suwanto1,2* 1Graduate School of Biotechnology, Bogor Agricultural University, Bogor 16680, Indonesia 2Biotechnology Research and Development, PT Wilmar Benih Indonesia, Bekasi 17530, Indonesia Received: December 8, 2018 Revised: May 1, 2019 Lipases are industrial enzymes that catalyze both triglyceride hydrolysis and ester synthesis. Accepted: May 1, 2019 The overexpression of lipase genes is considered one of the best approaches to increase the First published online enzymatic production for industrial applications. Subfamily I.2. lipases require a chaperone or May 14, 2019 foldase in order to become a fully-activated enzyme. The goal of this research was to isolate, *Corresponding author clone, and co-express genes that encode lipase and foldase from Burkholderia territorii GP3, a Phone: +62-812-9973-680; lipolytic bacterial isolate obtained from Mount Papandayan soil via growth on Soil Extract E-mail: [email protected] Rhodamine Agar. Genes that encode for lipase (lipBT) and foldase (lifBT) were successfully cloned from this isolate and co-expressed in the E. coli BL21 background. The highest expression was shown in E. coli BL21 (DE3) pLysS, using pET15b expression vector. LipBT was particulary unique as it showed highest activity with optimum temperature of 80°C at pH 11.0. The optimum substrate for enzyme activity was C10, which is highly stable in methanol solvent. -

Iron Transport Strategies of the Genus Burkholderia
Zurich Open Repository and Archive University of Zurich Main Library Strickhofstrasse 39 CH-8057 Zurich www.zora.uzh.ch Year: 2015 Iron transport strategies of the genus Burkholderia Mathew, Anugraha Posted at the Zurich Open Repository and Archive, University of Zurich ZORA URL: https://doi.org/10.5167/uzh-113412 Dissertation Published Version Originally published at: Mathew, Anugraha. Iron transport strategies of the genus Burkholderia. 2015, University of Zurich, Faculty of Science. Iron transport strategies of the genus Burkholderia Dissertation zur Erlangung der naturwissenschaftlichen Doktorwürde (Dr. sc. nat.) vorgelegt der Mathematisch-naturwissenschaftlichen Fakultät der Universität Zürich von Anugraha Mathew aus Indien Promotionskomitee Prof. Dr. Leo Eberl (Vorsitz) Prof. Dr. Jakob Pernthaler Dr. Aurelien carlier Zürich, 2015 2 Table of Contents Summary .............................................................................................................. 7 Zusammenfassung ................................................................................................ 9 Abbreviations ..................................................................................................... 11 Chapter 1: Introduction ....................................................................................... 14 1.1.Role and properties of iron in bacteria ...................................................................... 14 1.2.Iron transport mechanisms in bacteria ..................................................................... -

Comisión De Investigaciones Científicas CIC PBA, Ministerio De Producción, Ciencia Y Tecnología De La Provincia De Buenos Aires
“2014 – Año de Homenaje al Almirante Guillermo Brown, en el Bicentenario del Combate Naval de Montevideo” CARRERA DEL INVESTIGADOR CIENTÍFICO Y TECNOLÓGICO Informe Científico1 PERIODO 2: 2013-2015 1. DATOS PERSONALES APELLIDO: BOSCH NOMBRES: MARÍA ALEJANDRA NIEVES Dirección Particular: Calle: Localidad: City Bell CP: 1896 Dirección electrónica (donde desea recibir información, que no sea “Hotmail”): [email protected] 2. TEMA DE INVESTIGACION A) Caracterización molecular y fenotípica de organismos del complejo Burkholderia cepacia recuperados de infecciones pulmonares crónicas en pacientes con fibrosis quística. B) Empleo de métodos espectroscópicos vibracionales aplicados a la caracterización de sistemas biológicos. 3. DATOS RELATIVOS A INGRESO Y PROMOCIONES EN LA CARRERA INGRESO: Categoría: Investigador Adjunto sin Director Fecha: 06/04/2011 ACTUAL: Categoría: Investigador Adjunto sin Director desde fecha: 06/04/2011 4. INSTITUCION DONDE DESARROLLA LA TAREA Universidad y/o Centro: CINDEFI-CONICET-CCT La Plata, Facultad Ciencias Exactas Facultad: Ciencias Exactas, Universidad Nacional de La Plata Departamento: Química Cátedra: Biofilms microbianos / Ingeniería Bioquímica Otros: Área de Biotecnología Dirección: Calle: 50 Nº: 227 entre 115 y 116 Localidad: La Plata CP: 1900 Tel: 221 483 3794 Cargo que ocupa: Profesor Adjunto D. E. 5. DIRECTOR DE TRABAJOS. (En el caso que corresponda) 1 Art. 11; Inc. “e”; Ley 9688 (Carrera del Investigador Científico y Tecnológico). 2 El informe deberá referenciar a años calendarios completos. Ej.: en el año 2014 deberá informar sobre la actividad del período 1°-01-2012 al 31-12-2013, para las presentaciones bianuales. Formulario Informe Científico-Tecnológico 1 “2014 – Año de Homenaje al Almirante Guillermo Brown, en el Bicentenario del Combate Naval de Montevideo” Apellido y Nombres: Dirección Particular: Calle: Nº: Localidad: CP: Tel: Dirección electrónica: .................................................... -

Volcanic Soils As Sources of Novel CO-Oxidizing Paraburkholderia and Burkholderia: Paraburkholderia Hiiakae Sp
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Frontiers - Publisher Connector ORIGINAL RESEARCH published: 21 February 2017 doi: 10.3389/fmicb.2017.00207 Volcanic Soils as Sources of Novel CO-Oxidizing Paraburkholderia and Burkholderia: Paraburkholderia hiiakae sp. nov., Paraburkholderia metrosideri sp. nov., Paraburkholderia paradisi sp. nov., Edited by: Paraburkholderia peleae sp. nov., Svetlana N. Dedysh, Winogradsky Institute of Microbiology and Burkholderia alpina sp. nov. a (RAS), Russia Reviewed by: Member of the Burkholderia cepacia Dimitry Y. Sorokin, The Federal Research Centre Complex “Fundamentals of Biotechnology” of the Russian Academy of Sciences, 1, 2 1 Russia Carolyn F. Weber and Gary M. King * Philippe Constant, 1 Department of Biological Sciences, Louisiana State University, Baton Rouge, LA, USA, 2 College of Health Sciences, Des INRS-Institut Armand-Frappier, Moines University, Des Moines, IA, USA Canada Paulina Estrada De Los Santos, Instituto Politécnico Nacional, Mexico Previous studies showed that members of the Burkholderiales were important in *Correspondence: the succession of aerobic, molybdenum-dependent CO oxidizing-bacteria on volcanic Gary M. King [email protected] soils. During these studies, four isolates were obtained from Kilauea Volcano (Hawai‘i, USA); one strain was isolated from Pico de Orizaba (Mexico) during a separate Specialty section: study. Based on 16S rRNA gene sequence similarities, the Pico de Orizaba isolate This article was submitted to Terrestrial Microbiology, and the isolates from Kilauea Volcano were provisionally assigned to the genera a section of the journal Burkholderia and Paraburkholderia, respectively. Each of the isolates possessed a form Frontiers in Microbiology I coxL gene that encoded the catalytic subunit of carbon monoxide dehydrogenase Received: 09 December 2016 (CODH); none of the most closely related type strains possessed coxL or oxidized Accepted: 30 January 2017 Published: 21 February 2017 CO. -

Targeting the Burkholderia Cepacia Complex
viruses Review Advances in Phage Therapy: Targeting the Burkholderia cepacia Complex Philip Lauman and Jonathan J. Dennis * Department of Biological Sciences, University of Alberta, Edmonton, AB T6G 2E9, Canada; [email protected] * Correspondence: [email protected]; Tel.: +1-780-492-2529 Abstract: The increasing prevalence and worldwide distribution of multidrug-resistant bacterial pathogens is an imminent danger to public health and threatens virtually all aspects of modern medicine. Particularly concerning, yet insufficiently addressed, are the members of the Burkholderia cepacia complex (Bcc), a group of at least twenty opportunistic, hospital-transmitted, and notoriously drug-resistant species, which infect and cause morbidity in patients who are immunocompromised and those afflicted with chronic illnesses, including cystic fibrosis (CF) and chronic granulomatous disease (CGD). One potential solution to the antimicrobial resistance crisis is phage therapy—the use of phages for the treatment of bacterial infections. Although phage therapy has a long and somewhat checkered history, an impressive volume of modern research has been amassed in the past decades to show that when applied through specific, scientifically supported treatment strategies, phage therapy is highly efficacious and is a promising avenue against drug-resistant and difficult-to-treat pathogens, such as the Bcc. In this review, we discuss the clinical significance of the Bcc, the advantages of phage therapy, and the theoretical and clinical advancements made in phage therapy in general over the past decades, and apply these concepts specifically to the nascent, but growing and rapidly developing, field of Bcc phage therapy. Keywords: Burkholderia cepacia complex (Bcc); bacteria; pathogenesis; antibiotic resistance; bacterio- phages; phages; phage therapy; phage therapy treatment strategies; Bcc phage therapy Citation: Lauman, P.; Dennis, J.J. -

Characterization of Antimicrobial-Producing Beneficial Bacteria Isolated from Huanglongbing Escape Citrus Trees
fmicb-08-02415 December 5, 2017 Time: 16:45 # 1 ORIGINAL RESEARCH published: 07 December 2017 doi: 10.3389/fmicb.2017.02415 Characterization of Antimicrobial-Producing Beneficial Bacteria Isolated from Huanglongbing Escape Citrus Trees Nadia Riera1, Utpal Handique1, Yunzeng Zhang1, Megan M. Dewdney2 and Nian Wang1* 1 Citrus Research and Education Center, Department of Microbiology and Cell Science, Institute of Food and Agricultural Sciences, University of Florida, Lake Alfred, FL, United States, 2 Citrus Research and Education Center, Department of Plant Pathology, Institute of Food and Agricultural Sciences, University of Florida, Lake Alfred, FL, United States The microbiome associated with crop plants has a strong impact on their health and productivity. Candidatus Liberibacter asiaticus (Las), the bacterial pathogen responsible for Huanglongbing (HLB) disease, lives inside the phloem of citrus plants Edited by: Hua Lu, including the root system. It has been suggested that Las negatively affects citrus University of Maryland, Baltimore, microbiome. On the other hand, members of citrus microbiome also influence the United States interaction between Las and citrus. Here, we report the isolation and characterization Reviewed by: Huang Lili, of multiple putative beneficial bacteria from healthy citrus rhizosphere. Firstly, six Northwest A&F University, China bacterial strains showing antibacterial activity against two bacteria closely related to Zhengqing Fu, Las: Agrobacterium tumefaciens and Sinorhizobium meliloti were selected. Among University of South Carolina, United States them, Burkholderia metallica strain A53 and Burkholderia territorii strain A63 are *Correspondence: within the b-proteobacteria class, whereas Pseudomonas granadensis strain 100 and Nian Wang Pseudomonas geniculata strain 95 are within the g-proteobacteria class. -

Molecular Characterisation and Plant-Growth Promotion
MOLECULAR CHARACTERISATION AND PLANT-GROWTH PROMOTION POTENTIAL OF PHOSPHATE SOLUBILISING BACTERIA FROM ROOTS OF SELECTED CROPS AROUND MOROGORO MUNICIPALITY, TANZANIA STEPHEN, GERISON SADDICK A DISSERTATION SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE IN SOIL SCIENCE AND LAND MANAGEMENT OF SOKOINE UNIVERSITY OF AGRICULTURE. MOROGORO, TANZANIA. 2020 2 EXTENDED ABSTRACT Soil infertility is reported to be among the most limiting factors for crop production and yield. Despite being abundant in most soils, only a small proportion of phosphorus is readily available to plants due to its high reactivity with soil constituents and slow release from phosphate compounds. Phosphate solubilising bacteria (PSB) play an important role in phosphorus nutrition. These microbes can solubilise various insoluble phosphate compounds through different mechanisms including production of organic and inorganic acids, production of chelating substances and ammonium assimilation, thus enriching soluble phosphorus into soil solution for plant uptake. Use of phosphate solubilising bacteria in agriculture has been reported to increase crop yield in different crops including maize (Zea mays L.). Other than phosphate solubilisation, PSB can also solubilise micronutrients including zinc (Zn) and iron (Fe). PSB are also known to produce various plant growth promoting substances such as indole acetic acid (IAA) and siderophore, which are important for crop growth and development. This study therefore aimed at evaluating the plant growth promotion potential of phosphate solubilising microorganisms by, in addition to phosphate solubilisation, looking at their potential for zinc solubilisation, siderophore and IAA production and plant growth promotion in general. Purified colonies of bacteria from 19 native PSB isolated from selected field and garden crops grown around Morogoro municipality, Tanzania, were found to be strong phosphate solubilisers, hence were selected for further studies. -

DISSERTAÇÃO Josineide Neri Monteiro.Pdf
UNIVERSIDADE FEDERAL DE PERNAMBUCO CENTRO DE TECNOLOGIA E GEOCIÊNCIAS DEPARTAMENTO DE ENGENHARIA BIOMÉDICA PROGRAMA DE PÓS-GRADUAÇÃO EM ENGENHARIA BIOMÉDICA JOSINEIDE NERI MONTEIRO IDENTIFICAÇÃO DE BACTÉRIAS DO COMPLEXO Burkholderia cepacia ATRAVÉS DE UTILIZAÇÃO DE FERRAMENTAS COMPUTACIONAIS RECIFE 2017 JOSINEIDE NERI MONTEIRO IDENTIFICAÇÃO DE BACTÉRIAS DO COMPLEXO Burkholderia cepacia ATRAVÉS DE UTILIZAÇÃO DE FERRAMENTAS COMPUTACIONAIS Dissertação apresentada ao Programa de Pós-Graduação em Engenharia Biomédica (PPGEB), da Universidade Federal de Pernambuco (UFPE), como requisito parcial para a obtenção do título de Mestre em Engenharia Biomédica. Área de concentração: Computação Biomédica Linha de pesquisa: Inteligência Artificial e Sistemas Inteligentes ORIENTADOR: Ricardo Yara RECIFE 2017 Catalogação na fonte Bibliotecária Margareth Malta, CRB-4 / 1198 M775i Monteiro, Josineide Neri. Identificação de bactérias do complexo Burkholderia cepacia através de utilização de ferramentas computacionais / Josineide Neri Monteiro. - 2017. 75 folhas, il., gráfs., tabs. Orientador: Prof. Dr. Ricardo Yara. Dissertação (Mestrado) – Universidade Federal de Pernambuco. CTG. Programa de Pós-Graduação em Engenharia Biomédica, 2017. Inclui Referências e Apêndices. 1. Engenharia Biomédica. 2. Burkholderia. 3. Bioinformática. 4. Alinhamento genético. 5. Algoritmos genéticos. 6. Taxonomia. I. Yara, Ricardo. (Orientador). II. Título. UFPE 610.28 CDD (22. ed.) BCTG/2017-299 JOSINEIDE NERI MONTEIRO IDENTIFICAÇÃO DE BACTÉRIAS DO COMPLEXO Burkholderia Cepacia ATRAVÉS DE UTILIZAÇÃO DE FERRAMENTAS COMPUTACIONAIS Esta dissertação foi julgada adequada para a obtenção do título de Mestre em Engenharia Biomédica e aprovada em sua forma final pelo Orientador e pela Banca Examinadora. Orientador: ____________________________________ Prof. Dr. Ricardo Yara (Doutor pela Universidade de São Paulo – São Paulo , Brasil) Banca Examinadora: Prof. Dr. Ricardo Yara, UFPE Doutor pela Universidade de São Paulo – São Paulo, Brasil Prof. -
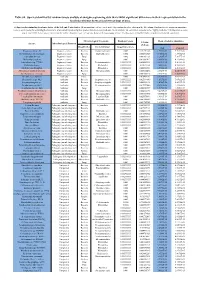
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

Molecular Study of the Burkholderia Cenocepacia Division Cell Wall Operon and Ftsz Interactome As Targets for New Drugs
Università degli Studi di Pavia Dipartimento di Biologia e Biotecnologie “L. Spallanzani” Molecular study of the Burkholderia cenocepacia division cell wall operon and FtsZ interactome as targets for new drugs Gabriele Trespidi Dottorato di Ricerca in Genetica, Biologia Molecolare e Cellulare XXXIII Ciclo – A.A. 2017-2020 Università degli Studi di Pavia Dipartimento di Biologia e Biotecnologie “L. Spallanzani” Molecular study of the Burkholderia cenocepacia division cell wall operon and FtsZ interactome as targets for new drugs Gabriele Trespidi Supervised by Prof. Edda De Rossi Dottorato di Ricerca in Genetica, Biologia Molecolare e Cellulare XXXIII Ciclo – A.A. 2017-2020 Cover image: representation of the Escherichia coli division machinery. Created by Ornella Trespidi A tutta la mia famiglia e in particolare a Diletta Table of contents Abstract ..................................................................................................... 1 Abbreviations ............................................................................................ 3 1. Introduction ........................................................................................... 5 1.1 Cystic fibrosis 5 1.1.1 CFTR defects in CF 5 1.1.2 Pathophysiology of CF 7 1.1.3 CFTR modulator therapies 11 1.2 CF-associated lung infections 12 1.2.1 Pseudomonas aeruginosa 13 1.2.2 Staphylococcus aureus 14 1.2.3 Stenotrophomonas maltophilia 15 1.2.4 Achromobacter xylosoxidans 16 1.2.5 Non-tuberculous mycobacteria 17 1.3 The genus Burkholderia 18 1.3.1 Burkholderia cepacia complex 20 1.3.2 Bcc species as CF pathogens 22 1.4 Burkholderia cenocepacia 23 1.4.1 B. cenocepacia epidemiology 23 1.4.2 B. cenocepacia genome 25 1.4.3 B. cenocepacia pathogenicity and virulence factors 26 1.4.3.1 Quorum sensing 29 1.4.3.2 Biofilm 32 1.4.4 B. -

The Organization of the Quorum Sensing Luxi/R Family Genes in Burkholderia
Int. J. Mol. Sci. 2013, 14, 13727-13747; doi:10.3390/ijms140713727 OPEN ACCESS International Journal of Molecular Sciences ISSN 1422-0067 www.mdpi.com/journal/ijms Article The Organization of the Quorum Sensing luxI/R Family Genes in Burkholderia Kumari Sonal Choudhary 1, Sanjarbek Hudaiberdiev 1, Zsolt Gelencsér 2, Bruna Gonçalves Coutinho 1,3, Vittorio Venturi 1,* and Sándor Pongor 1,2,* 1 International Centre for Genetic Engineering and Biotechnology (ICGEB), Padriciano 99, Trieste 32149, Italy; E-Mails: [email protected] (K.S.C.); [email protected] (S.H.); [email protected] (B.G.C.) 2 Faculty of Information Technology, PázmányPéter Catholic University, Práter u. 50/a, Budapest 1083, Hungary; E-Mail: [email protected] 3 The Capes Foundation, Ministry of Education of Brazil, Cx postal 250, Brasilia, DF 70.040-020, Brazil * Authors to whom correspondence should be addressed; E-Mails: [email protected] (V.V.); [email protected] (S.P.); Tel.: +39-40-375-7300 (S.P.); Fax: +39-40-226-555 (S.P.). Received: 30 May 2013; in revised form: 20 June 2013 / Accepted: 24 June 2013 / Published: 2 July 2013 Abstract: Members of the Burkholderia genus of Proteobacteria are capable of living freely in the environment and can also colonize human, animal and plant hosts. Certain members are considered to be clinically important from both medical and veterinary perspectives and furthermore may be important modulators of the rhizosphere. Quorum sensing via N-acyl homoserine lactone signals (AHL QS) is present in almost all Burkholderia species and is thought to play important roles in lifestyle changes such as colonization and niche invasion.