Borinine (Borabenzene): Its Structure and Vibrational Spectrum
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Communications to the Editor 6499
Communications to the Editor 6499 Nbenzyloxycarbonyl benzyl ester 7. 368 (1971). (17) D. M. Brunwin and G. Lowe, J. Chem. SOC., Perkin Trans. 1, 1321 (34) Computer programs used for these calculations were the X-ray 1972 (1973). system, Technical Report TR-192, Computer Science Center, University (18) R. B. Woodward, "Recent Advances in the Chemistry of p-Lactam Anti- of Maryland, College Park, Md. biotics", Chem. SOC., Spec. Pub/., NO. 28, 167-180 (1977). (35) C. K. Johnson, ORTEP, Oak Ridge National Laboratory, Report ORNL- (19) D. B. Bryan, R. F. Hall, K. G. Holden, W. F. Huffman, and J. G. Gleason, J. 3794. Am. Chem. SOC., 99,2353 (1977). (36) R. M. Sweet in "Cephalosporins and Penicillins", E. H. Flynn, Ed.. Academic (20)T. W. Doyle, J. L. Dowlas, B. Beleau. J. Mennier. and B. Luh. Can. J. Chem., Press, New York, N.Y., 1972, p. 297. 55, 2873 (1977). - (37) X-ray data from H. E. Applegate, J. E. Dolfini, M. S.Puar, W. A. Slusarchyk. (21) F DiNinno. T. R. Beattie, and B. G. Christensen, J. Org. Chem, 42, 1960 and B. Toeplitz, J. Org. Chem., 39, 2794 (1974). (19771\.- ,. (38) Nonfused 4-thioazetidin-2-one intermediates in the synthesis of 6P-ac- (22) Related reductions in penicillin derivatives give predominantly Cis-sJDsli- ylaminopenem esters, e.g., 4~-acetylthio-3P-phenoxyacetylaminoazeti- tded products: J. C. Sheehan and Y. S. Lo, J. Org. Chem., 38, 3227 (19731; din-2-one or terf-butyl 2-(4@-acetyithio-2-oxo-3~-phenoxyacetylamino- F. DiNinno, unpublisned resdts. l-azetidinyl)-2-hydroxyacetate in CH~CIP,show the p-lactam band at 1782 (23) K. -

Icr Annual Report 2005
ISSN 1342-0321 IAREFM ICR ANNUAL REPORT 2005 Volume 12 Institute for Chemical Research Kyoto University ICR ANNUAL REPORT 2005 (Volume 12) - ISSN 1342-0321 - This Annual Report covers from 1 January to 31 December 2005 Editors: Professor: OZAWA, Fumiyuki Professor: KANAYA, Toshiji Associate Professor: NISHIDA, Koji Associate Professor: GOTO, Susumu Editorial Staff: Public Relations Section: TSUGE, Aya GENMA, Mieko KOTANI, Masayo Published and Distributed by: Institute for Chemical Research (ICR), Kyoto University Copyright 2006 Institute for Chemical Research, Kyoto University Enquiries about copyright and reproduction should be addressed to: ICR Annual Report Committee, Institute for Chemical Research, Kyoto University Note: ICR Annual Report available from the ICR Office, Institute for Chemical Research, Kyoto University, Gokasho, Uji, Kyoto 611-0011, Japan Tel: +81-(0)774-38-3344 Fax: +81-(0)774-38-3014 E-mail [email protected] URL http://www.kuicr.kyoto-u.ac.jp/index.html Uji Library, Kyoto University Tel: +81-(0)774-38-3011 Fax: +81-(0)774-38-4370 E-mail [email protected] URL http://lib.kuicr.kyoto-u.ac.jp/homepage/english/homepageeng.html Printed by: Nakanishi Printing Co., Ltd. Ogawa Higashi-iru, Shimodachiuri, Kamigyo-ku, Kyoto 602-8048, Japan TEL:+81-(0)75-441-3155 FAX:+81-(0)75-417-2050 ICR AN NU AL REPORT 2005 Institute for Chemical Research Kyoto University Volume 12 Pref ace Institute for Chemical Research at Kyoto University has Moreover, we are encouraging community education to celebrated its 79th anniversary in October 2005. Initially, communicate the signifi cance and appeal of cutting-edge the size of the Institute was not substantial and it contained research through our “Chemical Research for High School only a limited number of laboratories, but growth soon ac- Students” and “Open Campus” programs. -

On the Impact of Excited State Antiaromaticity Relief in a Fundamental
On the Impact of Excited State Antiaromaticity Relief in a Fundamental Benzene Photoreaction Leading to Substituted Bicyclo[3.1.0]hexenes Tomáš Slanina,1,2 Rabia Ayub,1,‡ Josene Toldo,1,‡ Johan Sundell,3 Wangchuk Rabten,1 Marco Nicaso,1 Igor Alabugin,4 Ignacio Fdez. Galván,5 Arvind K. Gupta,1 Roland Lindh,5,6 Andreas Orthaber,1 Richard J. Lewis,7 Gunnar Grönberg,7 Joakim Bergman3,* and Henrik Ottosson1,* 1 Department of Chemistry – Ångström Laboratory, Uppsala University, SE-751 20, Uppsala, Sweden. 2 Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo námĕstí 2, 16610 Prague 6, Czech Republic 3 Medicinal Chemistry, Research and Early Development Cardiovascular, Renal and Metabolism, BioPharmaceuticals R&D, AstraZeneca, Gothenburg, Sweden. 4 Department of Chemistry and Biochemistry, Florida State University, Tallahassee, FL 32306-4390, USA, 5 Department of Chemistry – BMC, Uppsala University, 751 23, Uppsala, Sweden, 6 Uppsala Center for Computational 7 Chemistry – UC3, Uppsala University, 751 23, Uppsala Sweden, Medicinal Chemistry, Research and Early Development Respiratory, Inflammation and Autoimmune, BioPharmaceutical R&D, AstraZeneca Gothenburg, Sweden. ‡ These authors contributed equally. Table of contents 1. Materials and Methods ..................................................................................................................... 3 1.1 General information ..................................................................................................................... -
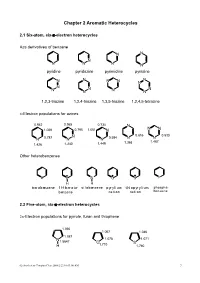
Chapter 2 Aromatic Heterocycles
Chapter 2 Aromatic Heterocycles 2.1 Six-atom, six-π-electron heterocycles Aza derivatives of benzene N N N N N N N pyridine pyridazine pyrimidine pyrazine N N N N N N N N N N N N N 1,2,3-triazine 1,2,4-triazine 1,3,5-triazine 1,2,4,5-tetrazine π-Electron populations for azines 0.942 0.965 0.734 N 1.029 0.795 1.051 N N N N 0.816 0.533 N 0.787 N N 0.584 N N 1.368 1.467 1.426 1.240 1.449 Other heterobenzenes B B Si O S P H H borabenzene 1H-borata- silabenzene pyrylium thiopyrylium phospha- benzene cation cation benzene 2.2 Five-atom, six-π-electron heterocycles 2π-Electron populations for pyrrole, furan and thiophene 1.090 1.067 1.046 1.087 N 1.078 1.071 1.5647 O 1.710 S H 1.760 02-Aro-hetero Chinpiao Chen 2008/2/22 10:03:00 AM 7 Aromatic five-membered heterocycles with two or more heteroatoms N N N N N N N N N N N O S S H H H isothiazole oxazole thiazole 1H-pyrazole 1H-imidazole 1H-tetrazole 2.3 Benzo-fused ring systems Benzo-fused six-membered nitrogen heteroaromatics N N N N N N quinoline isoquinoline cinnoline quinazoline N N N N N N N N N N quinoxaline phthalazine 1,2,3-benzotriazine 1,2,4-benzotriazine Five-membered benzo-fused heterocycles NH N O S H indole benzofuran benzo[b]thiophene isoindole N N O N N N O N H H isobenzofuran benzimidazole 1,2-benzisoxazole 1H-benzotriazole Other fused heterocycles N N N N N N N N N H N N phenanthridine pteridine purine quinolizinium cation indolizine 02-Aro-hetero Chinpiao Chen 2008/2/22 10:03:00 AM 8 2.4 Some criteria of aromaticity in heterocycles Bond lengths Typical lengths (Å) of single and -

Impact of Excited-State Antiaromaticity Relief in a Fundamental Benzene
This is an open access article published under a Creative Commons Attribution (CC-BY) License, which permits unrestricted use, distribution and reproduction in any medium, provided the author and source are cited. pubs.acs.org/JACS Article Impact of Excited-State Antiaromaticity Relief in a Fundamental Benzene Photoreaction Leading to Substituted Bicyclo[3.1.0]hexenes ∇ ∇ Tomaś̌Slanina, Rabia Ayub, Josene Toldo, Johan Sundell, Wangchuk Rabten, Marco Nicaso, Igor Alabugin, Ignacio Fdez. Galvan,́ Arvind K. Gupta, Roland Lindh, Andreas Orthaber, Richard J. Lewis, Gunnar Grönberg, Joakim Bergman,* and Henrik Ottosson* Cite This: J. Am. Chem. Soc. 2020, 142, 10942−10954 Read Online ACCESS Metrics & More Article Recommendations *sı Supporting Information ABSTRACT: Benzene exhibits a rich photochemistry which can provide access to complex molecular scaffolds that are difficult to access with reactions in the electronic ground state. While benzene is aromatic in its ground state, it is antiaromatic in its lowest ππ* excited states. Herein, we clarify to what extent relief of excited-state antiaromaticity (ESAA) triggers a fundamental benzene photoreaction: the photoinitiated nucleophilic addition of solvent to benzene in acidic media leading to substituted bicyclo[3.1.0]hex-2-enes. The reaction scope was probed experimentally, and it was found that silyl-substituted benzenes provide the most rapid access to bicyclo[3.1.0]hexene derivatives, formed as single isomers with three stereogenic centers in yields up to 75% in one step. Two major mechanism hypotheses, both involving ESAA relief, were explored through quantum chemical calculations and experiments. The first mechanism involves protonation of excited-state benzene and subsequent rearrange- ment to bicyclo[3.1.0]hexenium cation, trapped by a nucleophile, while the second involves photorearrangement of benzene to benzvalene followed by protonation and nucleophilic addition. -

Potentially Aromatic Metallocycles Kim K
Iowa State University From the SelectedWorks of Mark S. Gordon June, 1988 Potentially Aromatic Metallocycles Kim K. Baldridge Mark S. Gordon Available at: https://works.bepress.com/mark_gordon/69/ 4204 J. Am. Chern. Soc. 1988, 110, 4204-4208 Potentially Aromatic Metallocycles Kim K. Baldridge and MarkS. Gordon* Contribution from the Department of Chemistry, North Dakota State University, Fargo, North Dakota 58105. Received November 12, 1987 Abstract: Ab initio molecular orbital theory is used to characterize a series of metal-substituted benzene and cyclopentadiene structures, with the heteroatom taken from the block in the periodic table bounded by groups IV-VI and periods 2-5. Structures are predicted with the 3-21G* basis set and SCF wave functions. The calculated bond lengths and bond angles are in general within 0.04 A and 2°, respectively, of the available experimental values. As a measure of the de localization stabilization, !::.£and t::..H0 values for the appropriate bond separation and superhomodesmic reactions are calculated with 3-21 G* Hartree-Fock energies for these compounds and some smaller acyclic structures. I. Introduction Scheme I. Bond Separation Reactions a Group IV Considerable efforts have gone into the study of five- and X six-membered ring compounds. 1- 38 The theoretical and exper- 0 + SCH4 + XH4 (I) Baldridge, K. K.; Boatz, J. A.; Sakai, S.; Gordon, M.S. Annu. Rec. 2H 3C-CH3 + H3X-CH3 + 2H2C=CH2 + H2X=CH2 Phys. Chern .. in press. b Group V (2) Baldridge, K. K.; Gordon, M.S. Organometal/ics 1988, 7, 144-154. (3) Dewar, J. S.; Lo, D. H.; Ramsden, C. -

Syntheses of Elusive Unsaturated Silacycles Gary Thomas Burns Iowa State University
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1981 Syntheses of elusive unsaturated silacycles Gary Thomas Burns Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Burns, Gary Thomas, "Syntheses of elusive unsaturated silacycles " (1981). Retrospective Theses and Dissertations. 7402. https://lib.dr.iastate.edu/rtd/7402 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This was produced from a copy of a document sent to us for microfilming. While the most advanced technological means to photograph and reproduce this document have been used, the quality is heavily dependent upon the quality of the material submitted. The following explanation of techniques is provided to help you understand markings or notations which may appear on this reproduction. 1. The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting through an image and duplicating adjacent pages to assure you of complete continuity. 2. When an image on the film is obliterated with a round black mark it is an indication that the film inspector noticed either blurred copy because of movement during exposure, or duplicate copy. -

The Synthesis of Carbacyclic Silanes Via Organosilicon Reactive Intermediates Michael J
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1984 The synthesis of carbacyclic silanes via organosilicon reactive intermediates Michael J. Vuper Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Vuper, Michael J., "The synthesis of carbacyclic silanes via organosilicon reactive intermediates " (1984). Retrospective Theses and Dissertations. 7737. https://lib.dr.iastate.edu/rtd/7737 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS Tliis reproduction was made from a copy of a document sent to us for microfilming. While the most advanced technology has been used to photograph and reproduce this document, the quality of the reproduction is heavily dependent upon the quality of the material submitted. The following explanation of techniques is provided to help clarify markings or notations which may appear on this reproduction. 1. The sign or "target" for pages apparently lacking from the document photographed is "Missing Page(s)". If it was possible to obtain the missing page(s) or section, they are spliced into the film along with adjacent pages. This may have necessitated cutting through an image and duplicating adjacent pages to assure complete continuity. 2. When an image on the film is obliterated with a round black mark, it is an indication of either blurred copy because of movement during exposure, duplicate copy, or copyrighted materials that should not have been filmed. -

Hydrogenation of Small Aromatic Heterocycles at Low Temperatures
MNRAS 000,1–8 (2021) Preprint 25 May 2021 Compiled using MNRAS LATEX style file v3.0 Hydrogenation of small aromatic heterocycles at low temperatures April M. Miksch,1 Annalena Riffelt,1 Ricardo Oliveira,2 Johannes Kästner,1 and Germán Molpeceres,1¢ 1Institute for Theoretical Chemistry, University of Stuttgart, Stuttgart, Germany 2Chemistry Institute, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil Accepted XXX. Received YYY; in original form ZZZ ABSTRACT The recent wave of detections of interstellar aromatic molecules has sparked interest in the chemical behavior of aromatic molecules under astrophysical conditions. In most cases, these detections have been made through chemically related molecules, called proxies, that implicitly indicate the presence of a parent molecule. In this study, we present the results of the theoretical evaluation of the hydrogenation reactions of different aromatic molecules (benzene, pyridine, pyrrole, furan, thiophene, silaben- zene, and phosphorine). The viability of these reactions allows us to evaluate the resilience of these molecules to the most important reducing agent in the interstellar medium, the hydrogen atom (H). All significant reactions are exothermic and most of them present activation barriers, which are, in several cases, overcome by quantum tunneling. Instanton reaction rate constants are provided between 50 K and 500 K. For the most efficiently formed radicals, a second hydrogenation step has been studied. We propose that hydrogenated derivatives of furan, pyrrole, and specially 2,3-dihydropyrrole, 2,5-dihydropyrrole, 2,3-dihydrofuran, and 2,5-dihydrofuran are promising candidates for future interstellar detections. Key words: ISM: molecules – Molecular Data – Astrochemistry – methods: numerical 1 INTRODUCTION (Campbell et al. -

NBO Applications, 2008
NBO 2008 (Jan-Dec) - 910 references Compiled by Emily Wixson; Updated by Ariel Neff 4/16/13 Adalsteinsson, H.; Debusschere, B. J.; Long, K. R.; Najm, H. N. Components for atomistic-to-continuum multiscale modeling of flow in micro- and nanofluidic systems Scientific Programming, (16): 297-313 2008. Adcock, W.; Trout, N. A. Diastereofacial selectivity in some 4-substituted (X) 2-adamantyl derivatives: electronic versus steric effects Journal of Physical Organic Chemistry, (21): 68-72 2008. Agapito, F.; Nunes, P. A.; Costa Cabral, B. J.; Borges dos Santos, R. A.; Martinho Simoes, J. A. Energetic differences between the five- and six-membered ring hydrocarbons: Strain energies in the parent and radical molecules Journal of Organic Chemistry, (73): 6213-6223 2008. Aguilar-Castro, L.; Tlahuextl, M.; Mendoza-Huizar, L. H.; Tapia-Benavides, A. R.; Tlahuext, H. Hydrogen bond studies in substituted N-(2-hydroxyphenyl)-2-[(4- methylbenzenesulfonyl)amino]acetamides Arkivoc: 210-226 2008. Alajarin, M.; Cabrera, J.; Pastor, A.; Sanchez-Andrada, P.; Bautista, D. Polar hetero-Diels-alder reactions of 4-alkenylthiazoles with 1,2,4-triazoline-3,5-diones: An experimental and computational study Journal of Organic Chemistry, (73): 963-973 2008. Albertin, G.; Antoniutti, S.; Baldan, D.; Castro, J.; Garcia-Fontan, S. Preparation of benzyl azide complexes of iridium(III) Inorganic Chemistry, (47): 742-748 2008. Alcoba, D. R.; Ona, O. Determination of energies and electronic densities of functional groups according to partitionings in the physical space Journal of Physical Chemistry A, (112): 10023-10028 2008. Alia, J. D.; Vlaisavljevich, B. Prediction of molecular properties including symmetry from quantum-based molecular structural formulas, VIF Journal of Physical Chemistry A, (112): 9784-9795 2008. -

T. Don Tilley's Publications
Titanium-Germoxy Precursor Route to Germanium-Modified Epoxidation Catalysts with 387. Enhanced Activity. P. J. Cordeiro, P. Guillo, C. S. Spanjers, J. W. Chang, M. E. Fasulo, R. M. Rioux and T. D. Tilley, ACS Catal. 2013, 3, 2269. Photocatalytic Water Oxidation by Very Small Cobalt Domains on a Silica Surface. H. S. 386. Ahn, J. Yano and T. D. Tilley, Energy Environ. Sci. 2013, 6, 3080. Platinum-Catalyzed C-C Activation Induced by C-H Activation. M. Bowring, R. G. 385. Bergman and T. D. Tilley, J. Am. Chem. Soc. 2013, 135, 13121. Structural and mechanistic investigation of a cationic hydrogen-substituted ruthenium 384. silylene catalyst for alkene hydrosilation. M. Fasulo, M. C. Lipke and T. D. Tilley, Chem. Sci. 2013, 4, 3882. Water Oxidation Catalysis via Immobilization of the Dimanganese Complex [Mn2(μ- 383. O)2Cl(μ-O2CCH3)(bpy)2(H2O)](NO3)2 onto Silica. E. M. W. Rumberger, H. S. Ahn, A. T. Bell and T. D. Tilley, Dalton Trans. 2013, 42, 12238. The Osmium-Silicon Triple Bond: Synthesis and Reactivity of an Osmium Silylyne 382. Complex. P. G. Hayes, Z. Xu, C. Beddie, J. M. Keith, M. B. Hall and T. D. Tilley, J. Am. Chem. Soc. 2013, 135, 11780. Silane-Isocyanide Coupling Involving 1,1-Insertion of XylNC into the Si-H Bond of a σ- 381. Silane Ligand. M. C. Lipke and T. D. Tilley, J. Am. Chem. Soc. 2013, 135, 10298. A Remote Lewis Acid Trigger Dramatically Accelerates Biaryl Reductive Elimination from 380. a Platinum Complex. A. Liberman-Martin, R. G. Bergman and T. -

Borinine (Borabenzene): Its Structure and Vibrational Spectrum
Borinine (Borabenzene): Its Structure and Vibrational Spectrum. A Quantumchemical Study Gerhard Raabe, Wolfgang Schleker, Eberhard Heyne, and Jörg Fleischhauer Lehr- und Forschungsgebiet Theoretische Chemie der Rheinisch-Westfälischen Technischen Hochschule Aachen Z. Naturforsch. 42a, 352-360 (1987); received December 31, 1986 Recently we reported the results of some semiempirical and ab initio studies in which we compared the electronic structure of the hitherto unknown borinine with those of benzene and pyridine. The results of our calculations led us to the conclusion that the elusive nature of borabenzene is caused by its high reactivity, which might at least in part be due to the pronounced a acceptor properties of a low-lying er* molecular orbital. We now present the results of further ab initio and semiempirical (MNDO) investigations in which we performed full geometry optimizations for the molecule using two different basis sets (STO-3G, 4-31G) and also calculated the vibrational spectra of the 10B and "B isotopomeric borabenzene molecules at the 4-31 G level of ab initio theory and with the semiempirical MNDO method. The calculated vibrational spectrum might be helpful to the experimentalist in identifying the molecule, for example trapped in a rare gas matrix among the side products. The calculated orbital energies can be useful in identifying the molecule by means of its photoelectron spectrum. 1. Introduction Further it was found, that among its doubly occu- pied orbitals there are three of n symmetry. This Although a lot of borabenzene complexes are last result was confirmed by an STO-3G single known [1], and even though it was possible to trap point calculation using the MNDO optimized struc- the molecule with pyridine [2, 3] to our knowledge ture [4], If we accept three doubly occupied molec- no preparation and characterization of the unsub- ular orbitals of n symmetry in a planar perimeter as stituted neutral borinine* has been reported until a criterion for aromaticity, we have to consider the today.