Syntheses of Elusive Unsaturated Silacycles Gary Thomas Burns Iowa State University
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
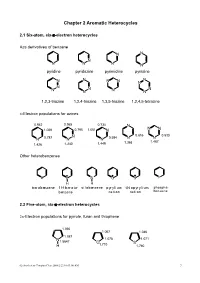
Chapter 2 Aromatic Heterocycles
Chapter 2 Aromatic Heterocycles 2.1 Six-atom, six-π-electron heterocycles Aza derivatives of benzene N N N N N N N pyridine pyridazine pyrimidine pyrazine N N N N N N N N N N N N N 1,2,3-triazine 1,2,4-triazine 1,3,5-triazine 1,2,4,5-tetrazine π-Electron populations for azines 0.942 0.965 0.734 N 1.029 0.795 1.051 N N N N 0.816 0.533 N 0.787 N N 0.584 N N 1.368 1.467 1.426 1.240 1.449 Other heterobenzenes B B Si O S P H H borabenzene 1H-borata- silabenzene pyrylium thiopyrylium phospha- benzene cation cation benzene 2.2 Five-atom, six-π-electron heterocycles 2π-Electron populations for pyrrole, furan and thiophene 1.090 1.067 1.046 1.087 N 1.078 1.071 1.5647 O 1.710 S H 1.760 02-Aro-hetero Chinpiao Chen 2008/2/22 10:03:00 AM 7 Aromatic five-membered heterocycles with two or more heteroatoms N N N N N N N N N N N O S S H H H isothiazole oxazole thiazole 1H-pyrazole 1H-imidazole 1H-tetrazole 2.3 Benzo-fused ring systems Benzo-fused six-membered nitrogen heteroaromatics N N N N N N quinoline isoquinoline cinnoline quinazoline N N N N N N N N N N quinoxaline phthalazine 1,2,3-benzotriazine 1,2,4-benzotriazine Five-membered benzo-fused heterocycles NH N O S H indole benzofuran benzo[b]thiophene isoindole N N O N N N O N H H isobenzofuran benzimidazole 1,2-benzisoxazole 1H-benzotriazole Other fused heterocycles N N N N N N N N N H N N phenanthridine pteridine purine quinolizinium cation indolizine 02-Aro-hetero Chinpiao Chen 2008/2/22 10:03:00 AM 8 2.4 Some criteria of aromaticity in heterocycles Bond lengths Typical lengths (Å) of single and -

NBO Applications, 2008
NBO 2008 (Jan-Dec) - 910 references Compiled by Emily Wixson; Updated by Ariel Neff 4/16/13 Adalsteinsson, H.; Debusschere, B. J.; Long, K. R.; Najm, H. N. Components for atomistic-to-continuum multiscale modeling of flow in micro- and nanofluidic systems Scientific Programming, (16): 297-313 2008. Adcock, W.; Trout, N. A. Diastereofacial selectivity in some 4-substituted (X) 2-adamantyl derivatives: electronic versus steric effects Journal of Physical Organic Chemistry, (21): 68-72 2008. Agapito, F.; Nunes, P. A.; Costa Cabral, B. J.; Borges dos Santos, R. A.; Martinho Simoes, J. A. Energetic differences between the five- and six-membered ring hydrocarbons: Strain energies in the parent and radical molecules Journal of Organic Chemistry, (73): 6213-6223 2008. Aguilar-Castro, L.; Tlahuextl, M.; Mendoza-Huizar, L. H.; Tapia-Benavides, A. R.; Tlahuext, H. Hydrogen bond studies in substituted N-(2-hydroxyphenyl)-2-[(4- methylbenzenesulfonyl)amino]acetamides Arkivoc: 210-226 2008. Alajarin, M.; Cabrera, J.; Pastor, A.; Sanchez-Andrada, P.; Bautista, D. Polar hetero-Diels-alder reactions of 4-alkenylthiazoles with 1,2,4-triazoline-3,5-diones: An experimental and computational study Journal of Organic Chemistry, (73): 963-973 2008. Albertin, G.; Antoniutti, S.; Baldan, D.; Castro, J.; Garcia-Fontan, S. Preparation of benzyl azide complexes of iridium(III) Inorganic Chemistry, (47): 742-748 2008. Alcoba, D. R.; Ona, O. Determination of energies and electronic densities of functional groups according to partitionings in the physical space Journal of Physical Chemistry A, (112): 10023-10028 2008. Alia, J. D.; Vlaisavljevich, B. Prediction of molecular properties including symmetry from quantum-based molecular structural formulas, VIF Journal of Physical Chemistry A, (112): 9784-9795 2008. -

Borinine (Borabenzene): Its Structure and Vibrational Spectrum
Borinine (Borabenzene): Its Structure and Vibrational Spectrum. A Quantumchemical Study Gerhard Raabe, Wolfgang Schleker, Eberhard Heyne, and Jörg Fleischhauer Lehr- und Forschungsgebiet Theoretische Chemie der Rheinisch-Westfälischen Technischen Hochschule Aachen Z. Naturforsch. 42a, 352-360 (1987); received December 31, 1986 Recently we reported the results of some semiempirical and ab initio studies in which we compared the electronic structure of the hitherto unknown borinine with those of benzene and pyridine. The results of our calculations led us to the conclusion that the elusive nature of borabenzene is caused by its high reactivity, which might at least in part be due to the pronounced a acceptor properties of a low-lying er* molecular orbital. We now present the results of further ab initio and semiempirical (MNDO) investigations in which we performed full geometry optimizations for the molecule using two different basis sets (STO-3G, 4-31G) and also calculated the vibrational spectra of the 10B and "B isotopomeric borabenzene molecules at the 4-31 G level of ab initio theory and with the semiempirical MNDO method. The calculated vibrational spectrum might be helpful to the experimentalist in identifying the molecule, for example trapped in a rare gas matrix among the side products. The calculated orbital energies can be useful in identifying the molecule by means of its photoelectron spectrum. 1. Introduction Further it was found, that among its doubly occu- pied orbitals there are three of n symmetry. This Although a lot of borabenzene complexes are last result was confirmed by an STO-3G single known [1], and even though it was possible to trap point calculation using the MNDO optimized struc- the molecule with pyridine [2, 3] to our knowledge ture [4], If we accept three doubly occupied molec- no preparation and characterization of the unsub- ular orbitals of n symmetry in a planar perimeter as stituted neutral borinine* has been reported until a criterion for aromaticity, we have to consider the today. -
Design, Synthesis and Mechanistic Studies of Boron and Phosphorus Heterocycles and Their Applications in Asymmetric Catalysis
Design, Synthesis and Mechanistic Studies of Boron and Phosphorus Heterocycles and Their Applications in Asymmetric Catalysis by Shuang Qiao B.A. Chemistry, Harvard University (1994) Submitted to the Department of Chemistry in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY IN ORGANIC CHEMISTRY at the Massachusetts Institute of Technology June 1999 ©Massachusetts Institute of Technology, 1999 All rights reserved Signature of Author Depar ent of Chemistry Vay 24, 1999 Certified by Gregory C. Fu Thesis Supervisor Accepted by Dietmar Seyferth Chairman, Departmental Committee on Graduate Students ASSACHUSETTS INSTITUTE OF TECHINOLOGY L "iAR7E -r. 9 - This doctoral thesis has been examined by a committee of the Department of Chemistry as follows: Professor Satoru Masamune I I --- -1 -116, - 011 tmp. - - - -r~s-~ Chairman Professor Gregory C. Fu Thesis Supervisor Professor Peter H. Seeberger 2 Design, Synthesis and Mechanistic Studies of Boron and Phosphorus Heterocycles and Their Applications in Asymmetric Catalysis by Shuang Qiao Submitted to the Department of Chemistry on May 24, 1999 in partial fulfillment of the requirements for the Degree of Doctor of Philosophy at the Massachusetts Institute of Technology ABSTRACT In Part I of this thesis, detailed synthetic and mechanistic studies of borabenzene and boratabenzene complexes are discussed. A comprehensive mechanistic study of nucleophilic aromatic substitution reactions of borabenzene-PMe3 complexes strongly favors an associative pathway, an addition-elimination mechanism, over all the other potential alternatives. Boratabenzenes are found to be capable of undergoing nucleophilic aromatic substitutions as well. Structurally interesting complexes have been synthesized and characterized. In Part II, a new approach to chiral phosphines in asymmetric catalysis is discussed. -

NBO 2016 – 2008 References Compiled by Ariel Andrea on 8/31/2018
NBO 2016 – 2008 references Compiled by Ariel Andrea on 8/31/2018 Aal, S. A. Reactivity of boron- and nitrogen-doped carbon nanotubes functionalized by (Pt, Eu) atoms toward O-2 and CO: A density functional study International Journal of Modern Physics C, (27) 2016. 10.1142/s0129183116500753 Abbat, S.; Bharatam, P. V. Electronic structure and conformational analysis of P218: An antimalarial drug candidate International Journal of Quantum Chemistry, (116): 1362-1369. 2016. 10.1002/qua.25189 Abbenseth, J.; Finger, M.; Wurtele, C.; Kasanmascheff, M.; Schneider, S. Coupling of terminal iridium nitrido complexes Inorganic Chemistry Frontiers, (3): 469-477. 2016. 10.1039/c5qi00267b Abboud, J. L. M.; Alkorta, I.; Davalos, J. Z.; Koppel, I. A.; Koppel, I.; Lenoir, D.; Martinez, S.; Mishima, M. The Thermodynamic Stability of Adamantylideneadamantane and Its Proton- and Electron-Exchanges. Comparison with Simple Alkenes Bulletin of the Chemical Society of Japan, (89): 762-769. 2016. 10.1246/bcsj.20160026 Abdalrazaq, S. M.; Cabir, B.; Gumus, S.; Agirtas, M. S. Synthesis of metallophthalocyanines with four oxy-2,2-diphenylacetic acid substituents and their structural and electronic properties Heterocyclic Communications, (22): 275-280. 2016. 10.1515/hc-2016-0120 Abdelmoulahi, H.; Ghalla, H.; Nasr, S.; Bahri, M.; Bellissent-Funel, M. C. Hydrogen-bond network in liquid ethylene glycol as studied by neutron scattering and DFT calculations Journal of Molecular Liquids, (220): 527-539. 2016. 10.1016/j.molliq.2016.04.111 Abdelmoulahi, H.; Ghalla, H.; Nasr, S.; Darpentigny, J.; Bellissent-Funel, M. C. Intermolecular associations in an equimolar formamide-water solution based on neutron scattering and DFT calculations Journal of Chemical Physics, (145) 2016. -

Benzene from Wikipedia, the Free Encyclopedia See Also: Benzole
Benzene From Wikipedia, the free encyclopedia See also: Benzole Benzene is an organic chemical compound with Benzene the molecular formula C6H6. Its molecule is composed of 6 carbon atoms joined in a ring, with 1 hydrogen atom attached to each carbon atom. Because its molecules contain only carbon and hydrogen atoms, benzene is classed as a hydrocarbon. Benzene is a natural constituent of crude oil, and is one of the most elementary petrochemicals. Benzene is an aromatic hydrocarbon and the second [n]-annulene ([6]-annulene), a cyclic hydrocarbon with a continuous pi bond. It is sometimes abbreviated Ph–H. Benzene is a colorless and highly flammable liquid with a sweet smell. It is mainly used as a precursor to heavy chemicals, such as ethylbenzene and cumene, which are produced on a billion kilogram scale. Because it has a high octane number, it is an important component of gasoline, composing a few percent of its mass. Most non-industrial applications have been limited by benzene's carcinogenicity. IUPAC name Contents benzene 1 History Systematic name 1.1 Discovery 1.2 Ring formula cyclohexa-1,3,5-triene 1.3 Early applications Other names 2 Structure 3 Benzene derivatives 1,3,5-cyclohexatriene 4 Production benzol 4.1 Catalytic reforming phene 4.2 Toluene hydrodealkylation 4.3 Toluene disproportionation Identifiers 4.4 Steam cracking CAS number 71-43-2 4.5 Other sources PubChem 241 5 Uses ChemSpider 236 5.1 Component of gasoline 6 Reactions UNII J64922108F 6.1 Sulfonation, chlorination, nitration EC number 200-753-7 6.2 Hydrogenation -

From Pyridine Adduct of Borabenzene to (In)Finite Graphene Architectures Functionalized with N→B Dative Bonds
From Pyridine Adduct of Borabenzene to (in)finite Graphene Architectures Functionalized with N!B Dative Bonds. Prototype Systems of Strong One and Two Photon Quantum Transitions Triggering Large Nonlinear Optical Responses Panaghiotis Karamanis, Nicolás Otero, Demetrios Xenides, Hassan Denawi, Marcos Mandado, Michel Rérat To cite this version: Panaghiotis Karamanis, Nicolás Otero, Demetrios Xenides, Hassan Denawi, Marcos Mandado, et al.. From Pyridine Adduct of Borabenzene to (in)finite Graphene Architectures Functionalized with N!B Dative Bonds. Prototype Systems of Strong One and Two Photon Quantum Transitions Triggering Large Nonlinear Optical Responses. Journal of Physical Chemistry C, American Chemical Society, 2020, 124, pp.21063-21074. 10.1021/acs.jpcc.0c05190. hal-02935781 HAL Id: hal-02935781 https://hal.archives-ouvertes.fr/hal-02935781 Submitted on 14 Dec 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. The Journal of Physical Chemistry This document is confidential and is proprietary to the American Chemical Society and its authors. Do not copy or disclose without written permission. If you have received this item in error, notify the sender and delete all copies. From Pyridine Adduct of Borabenzene to (in)finite Graphene Architectures Functionalized with N→B Dative Bonds. -

Borinine (Borabenzene): Its Structure and Vibrational Spectrum
Borinine (Borabenzene): Its Structure and Vibrational Spectrum. A Quantumchemical Study Gerhard Raabe, Wolfgang Schleker, Eberhard Heyne, and Jörg Fleischhauer Lehr- und Forschungsgebiet Theoretische Chemie der Rheinisch-Westfälischen Technischen Hochschule Aachen Z. Naturforsch. 42a, 352-360 (1987); received December 31, 1986 Recently we reported the results of some semiempirical and ab initio studies in which we compared the electronic structure of the hitherto unknown borinine with those of benzene and pyridine. The results of our calculations led us to the conclusion that the elusive nature of borabenzene is caused by its high reactivity, which might at least in part be due to the pronounced a acceptor properties of a low-lying er* molecular orbital. We now present the results of further ab initio and semiempirical (MNDO) investigations in which we performed full geometry optimizations for the molecule using two different basis sets (STO-3G, 4-31G) and also calculated the vibrational spectra of the 10B and "B isotopomeric borabenzene molecules at the 4-31 G level of ab initio theory and with the semiempirical MNDO method. The calculated vibrational spectrum might be helpful to the experimentalist in identifying the molecule, for example trapped in a rare gas matrix among the side products. The calculated orbital energies can be useful in identifying the molecule by means of its photoelectron spectrum. 1. Introduction Further it was found, that among its doubly occu- pied orbitals there are three of n symmetry. This Although a lot of borabenzene complexes are last result was confirmed by an STO-3G single known [1], and even though it was possible to trap point calculation using the MNDO optimized struc- the molecule with pyridine [2, 3] to our knowledge ture [4], If we accept three doubly occupied molec- no preparation and characterization of the unsub- ular orbitals of n symmetry in a planar perimeter as stituted neutral borinine* has been reported until a criterion for aromaticity, we have to consider the today. -

Studies Into the Effects of Hexa-Hapto Spectator Ligands on the Chemistry of Ruthenium
Studies into the Effects of Hexa-hapto Spectator Ligands on the Chemistry of Ruthenium A thesis presented to the University of London in partial fulfilment of the requirements for the degree of Doctor of Philosophy. Jonathan W. Steed. UCL UNIVERSITY COLLEGE LONDON, 1993. ProQuest Number: 10046152 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest. ProQuest 10046152 Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 To my mother, who brought me unscathed through the first eighteen years of my life and little damaged thereafter, and to Coreena, of course. ABSTRACT This thesis describes the results of investigations into the chemistry of ruthenium complexes of hexa-hapto spectator ligands. Two specific systems have been examined. Chapters 2 - 5 describe aspects of the chemistry of the ruthenium(IV) chloride bridged compound [ {Ru(T|^:T|^-CioHi6)Cl(p-Cl) which contains the r|^:r|^-Z?w(allyl) ligand 2,7-dimethylocta-2,6-diene-l,8-diyl, derived from the ruthenium trichloride mediated dimérisation of isoprene. In Chapters 6 and 7 the reactivity of arenes and cyclohexadienes is examined in ruthenium complexes of the polyaromatic T|^-spectator [2.2]paracyclophane. -

In Chemistry and Physics, the Inductive Effect Is an Experimentally
In chemistry and physics, the inductive effect is an experimentally observable effect of the transmission of charge through a chain of atoms in a molecule by electrostatic induction.[1] The net polar effect exerted by a substituent is a combination of this inductive effect and the mesomeric effect. The electron cloud in a σ-bond between two unlike atoms is not uniform and is slightly displaced towards the more electronegative of the two atoms. This causes a permanent state of bond polarization, where the more electronegative atom has a slight negative charge (δ–) and the other atom has a slight positive charge (δ+). If the electronegative atom is then joined to a chain of atoms, usually carbon, the positive charge is relayed to the other atoms in the chain. This is the electron-withdrawing inductive effect, also known as the − I effect. Some groups, such as the alkyl group are less electron-withdrawing than hydrogen and are therefore considered as electron-releasing. This is electron releasing character and is indicated by the + I effect. As the induced change in polarity is less than the original polarity, the inductive effect rapidly dies out, and is significant only over a short distance. The inductive effect is permanent but feeble, as it involves the shift of strongly held σ-bond electrons, and other stronger factors may overshadow this effect. The inductive effect may be caused by some molecules also. Relative inductive effects have been experimentally measured with reference to hydrogen. Inductive effects can be measured through the Hammett equation. Inductive Effect can also be used to determine whether a molecule is stable or unstable depending on the charge present on the atom under consideration and the type of groups bonded to it.