ROLE of Ca and SODIUM-CALCIUM EXCHANGER
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
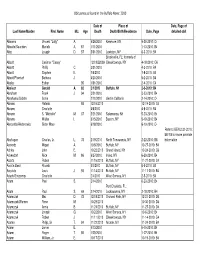
Obituaries Buffalo News 2010 by Name
Obituaries as found in the Buffalo News: 2010 Date of Place of Date, Page of Last Name/Maiden First Name M.I. Age Death Death/Birth/Residence Date, Page detailed obit Abbarno Vincent "Lolly" A. 9/26/2010 Kenmore, NY 9-30-2010: C4 Abbatte/Saunders Murielle A. 87 1/11/2010 1-13-2010: B4 Abbo Joseph D. 57 5/31/2010 Lewiston, NY 6-3-2010: B4 Brooksville, FL; formerly of Abbott Casimer "Casey" 12/19/22009 Cheektowaga, NY 4-18-2010: C6 Abbott Phillip C. 3/31/2010 4-3-2010: B4 Abbott Stephen E. 7/6/2010 7-8-2010: B4 Abbott/Pfoetsch Barbara J. 4/20/2010 5-2-2010: B4 Abeles Esther 95 1/31/2010 2-4-2010: C4 Abelson Gerald A. 82 2/1/2010 Buffalo, NY 2-3-2010: B4 Abraham Frank J. 94 3/21/2010 3-23-2010: B4 Abrahams/Gichtin Sonia 2/10/2010 died in California 2-14-2010: C4 Abramo Rafeala 93 12/16/2010 12-19-2010: C4 Abrams Charlotte 4/6/2010 4-8-2010: B4 Abrams S. "Michelle" M. 37 5/21/2010 Salamanca, NY 5-23-2010: B4 Abrams Walter I. 5/15/2010 Basom, NY 5-19-2010: B4 Abrosette/Aksterowicz Sister Mary 6/18/2010 6-19-2010: C4 Refer to BEN 2-21-2010: B6/7/8 for more possible Abshagen Charles, Jr. L. 73 2/19/2010 North Tonawanda, NY 2-22-2010: B8 information Acevedo Miguel A. 10/6/2010 Buffalo, NY 10-27-2010: B4 Achkar John E. -

The German Surname Atlas Project ± Computer-Based Surname Geography Kathrin Dräger Mirjam Schmuck Germany
Kathrin Dräger, Mirjam Schmuck, Germany 319 The German Surname Atlas Project ± Computer-Based Surname Geography Kathrin Dräger Mirjam Schmuck Germany Abstract The German Surname Atlas (Deutscher Familiennamenatlas, DFA) project is presented below. The surname maps are based on German fixed network telephone lines (in 2005) with German postal districts as graticules. In our project, we use this data to explore the areal variation in lexical (e.g., Schröder/Schneider µtailor¶) as well as phonological (e.g., Hauser/Häuser/Heuser) and morphological (e.g., patronyms such as Petersen/Peters/Peter) aspects of German surnames. German surnames emerged quite early on and preserve linguistic material which is up to 900 years old. This enables us to draw conclusions from today¶s areal distribution, e.g., on medieval dialect variation, writing traditions and cultural life. Containing not only German surnames but also foreign names, our huge database opens up possibilities for new areas of research, such as surnames and migration. Due to the close contact with Slavonic languages (original Slavonic population in the east, former eastern territories, migration), original Slavonic surnames make up the largest part of the foreign names (e.g., ±ski 16,386 types/293,474 tokens). Various adaptations from Slavonic to German and vice versa occurred. These included graphical (e.g., Dobschinski < Dobrzynski) as well as morphological adaptations (hybrid forms: e.g., Fuhrmanski) and folk-etymological reinterpretations (e.g., Rehsack < Czech Reåak). *** 1. The German surname system In the German speech area, people generally started to use an addition to their given names from the eleventh to the sixteenth century, some even later. -

1986 Surname
Surname Given Age Date Page Maiden Note Abegg Missel 88 15-Dec C-8 Abernathy Manuel 79 1-Jan C-5 Abner Tom 71 24-Jun B-7 Abraham Aloysius J. 77 24-Jul C-2 Abram Harold Glenn 75 14-Sep D-2 Abramson Frances L. 46 28-Dec C-7 Levine Ackerman Mary 79 16-Sep B-7 Adam Paul T. 76 10-Sep C-5 Adams Claude 78 20-Jan A-5 Adams Gloria L. 48 23-Sep D-6 Adams Irene 77 21-Aug C-3 Adams Martha 72 3-Nov C-1 Adank Gerald C. 65 1-Dec C-8 Veteran of World War II Adkins Johnnie Lee 78 13-Feb B-9 Adley Daisy A. 93 2-Sep D-7 Ahlborn Raymond W. 73 2-May C-1 Aird Gordon R. 77 21-Nov D-1 See article, p. D-1 Aitken Marion 70 1-Jul B-7 Aksentijevic Martha 56 17-Sep B-8 Alamillo Nora 75 4-Feb C-1 Albert Lester E. 65 30-Sep B-7 Albrecht Victoria A. 84 3-Jan A-7 Aldrin Raymond E. 71 11-Aug B-5 Aleksandrovic Ivan 76 11-Dec B-13 Aleksandrovic Jelena 79 2-Dec C-1 Ales Francis J. 69 31-Mar B-8 Alexander Janet 49 28-Jul C-1 Alexander Penny C. 60 24-Mar C-1 Alexander Terry 53 9-Dec C-1 Alexander Vera (Cook) 2-Dec C-1 Alexander William A. 50 27-Dec C-2 Alfaro Mark A. 21 5-Feb D-1 Alger Kenneth H. -

Inca Project No
1 FINAL REPORT INCA PROJECT NO. 223 THE BIOLOGICAL IMPORTANCE OF COPPER A Literature Review June, 1990 INCA PROJECT 223 Preface In 1973 the International Copper Research Association initiated a grant to review the literature dealing with the biological importance of copper in marine and estuarine environments. This was followed by a second review in 1978. It was then apparent that there was a very large number of publications concerning copper in the marine environment. As a result, an annual review was initiated. Reviews prior to 1984 considered copper only in marine and estuarine environments. However, events occurring on land and in freshwater were often mentioned because chemical and biological factors and processes pertinent to one environment could often be applied to the others. As a result, the review became larger, covering not only freshwater, saltwater and terrestrial environments but also agriculture and medicine. These broad reviews pointed out the broad application of concepts about the biological importance of copper. The present review includes literature for the period 1987-1988 although a number of earlier references are included and, where appropriate, a few appearing in 1989 have been used. Many of the earlier references are from Eastern Europe and Asia because this literature takes time to appear in the North American data review bases. References were obtained in major part through literature search programs available through the Woodward Biomedical Library at the University of British Columbia. Mr. Brian Moreton, the European INCA Director, kindly provided the metals section of the Marine Pollution Research Titles as a source of European as well as North American References. -

International Handbook of Occupational Therapy Interventions Ingrid Söderback Editor
International Handbook of Occupational Therapy Interventions Ingrid Söderback Editor International Handbook of Occupational Therapy Interventions Editor Ingrid Söderback Associate Professor in Occupational Therapy and Rehabilitation Karolinska Institute, Stockholm, Sweden and University Lecturer Emerita Department of Public Health and Caring Sciences Uppsala University, Uppsala, Sweden Home address: Sickla Kanalgata 31 nb SE 13165 Nacka E-mail: [email protected] ISBN: 978-0-387-75423-9 e-ISBN: 978-0-387-75424-6 DOI: 10.1007/978-0-387-75424-6 Springer Dordrecht Heidelberg London New York Library of Congress Control Number: 2009927464 © Springer Science+Business Media, LLC 2009 All rights reserved. This work may not be translated or copied in whole or in part without the written permission of the publisher (Springer Science+Business Media, LLC, 233 Spring Street, New York, NY 10013, USA), except for brief excerpts in connection with reviews or scholarly analysis. Use in connection with any form of information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed is forbidden. The use in this publication of trade names, trademarks, service marks, and similar terms, even if they are not identified as such, is not to be taken as an expression of opinion as to whether or not they are subject to proprietary rights. Printed on acid-free paper Springer is part of Springer Science+Business Media (www.springer.com) To be occupied is a fundamental right in every human being’s life. The occupational therapists’ main professional role is to encourage the clients’ occupational performances. To occupational therapy students, occupational therapists, and members of rehabilitation and health care teams, who are professionally working to improve their clients’ health and wellness, and to stakeholders responsible for the administration of occupational therapy, worldwide. -

Self Report English Version – Rafał Jakieła
Załącznik 3 Self report English version – Rafał Jakieła Warsaw, February 7, 2020 Dr Rafał Jakieła Laboratory of X-Ray and Electron Microscopy Research Group of X-ray Spectroscopy and Microanalysis Institute of Physics Polish Academy of Sciences Al. Lotników 32/46, 02-668 Warszawa e-mail: [email protected] SELF REPORT 1. Personal data: ...................................................................................................................... 2 2. Education and Degrees........................................................................................................ 2 3. Information on Previous Employment ................................................................................ 2 4. Description of the achievements, set out in art. 219 para 1 point 2 of the Act ................... 3 5. Scientific achievement being the subject of habilitation .................................................... 5 6. Bibliometric information .................................................................................................. 23 6.1 Total Number of Publications .................................................................................... 23 6.2 Total Impact Factor of publications ........................................................................... 23 6.3 Total citation number of publication ......................................................................... 23 6.4 Hirsch Index of publication ....................................................................................... 23 7. Presentation of -

Chamberlain University Commencement Ceremonies Post-Licensure & Graduate Degree Programs
Chamberlain University Commencement Ceremonies Post-Licensure & Graduate Degree Programs November 2020 Graduates Letter from the Chamberlain University President Dear Chamberlain Graduates, On behalf of the entire faculty and staff, it is my great pleasure to congratulate each one of you on your graduation. Today’s ceremony formally recognizes the culmination of your academic achievements and your commitment to the healthcare profession. Many thanks to the family and friends who have been your biggest supporters during your time at Chamberlain. We know how invaluable their encouragement has been in furthering your education and reaching your goals and dreams. We sincerely wish you all the best in your career and want you to be proud of your connection with Chamberlain University. It has been an honor to partner with you on your journey toward becoming an extraordinary healthcare professional. We hope that we have instilled a sense of pride in your profession, a desire for lifelong learning and a commitment to caring along the way. Congratulations on this tremendous milestone and thank you for your dedication to transforming healthcare in our local and global communities. Warm regards, Karen Cox, PhD, RN, FACHE, FAAN President Chamberlain University 2 Chamberlain University Post-Licensure and Graduate Commencement Ceremonies – November 2020 Table of Contents Letter from the Chamberlain University President 2 Keynote Speaker 4 Chamberlain University Commencement Ceremony 5 Chamberlain University Graduates College of Nursing Doctor of Nursing -

Basic Explanation of Polish Surname Endings RAYMOND JASTRZAB·FRIDAY, FEBRUARY 12, 2021· the POLISH NAME
Basic Explanation of Polish Surname Endings RAYMOND JASTRZAB·FRIDAY, FEBRUARY 12, 2021· THE POLISH NAME Polish names have two main elements: the imię, the first name, or given name; and the nazwisko, the last name, family name (surname). The usage of personal names in Poland is generally governed by civil law, church law, personal taste and family custom. The law requires a given name (imię) to indicate the person's gender. Almost all Polish female names end in a vowel -a, and most male names end in a consonant or a vowel other than a. There are, however, a few male names that end in a, which are very old and uncommon, such as Barnaba, Bonawentura, Boryna, Jarema, Kosma, Kuba (a diminutive of Jakub) and Saba. Maria is a female name that can be used also as a middle (second) name for males. Since the High Middle Ages, Polish-sounding surnames ending with the masculine - ski suffix (including -cki and -dzki, and the corresponding feminine suffix -ska/- https://www.facebook.com/notes/eastern-europe-genealogy-research-community/basic-explanation- of-polish-surname-endings/1090999978046463/ April 4, 2021 cka/-dzka), were associated with the nobility (Polish szlachta), which alone, in the early years, had such suffix distinctions. However, they are widely popular today. Minor regional spelling differences also exist depending on whether the surname originated in Polish, Czechor Slovak (-sky/-ský). THE Imię (given name) A child in Poland is usually given one or two names; Polish registry offices do not register more than two. Among Catholics, who form the vast majority of the population, it is customary to adopt the name of a saint as an informal, third given name at confirmation, however, this does not have any legal effect. -

Amelia Earhart Memorial Scholarship Winners 1941-Present, by Name
Amelia Earhart Scholarship Winners, 1941 to 2021, by Name Country Year Surname 1stName MName PrevName SCH Desc Other (Non US) Sect Chapter 2014 Abraham Laura Heli Add-On Also 2016 Mid-Atl Old Dominion 2016 Abraham Laura Multi-Engine Comm Also 2014 Mid-Atl Old Dominion 1983 Alexander Linda "Mike" ATP Airlines-Continental SCS Houston 2002 Allen Mary CFI-Multi-Engine Mid-Atl Hampton Roads 1994 Allinson Gail Lynne Comm NCS Chicago Area 1992 Ambrose Evelyn "Evie" Multi-Engine NCS Chicago Area 2019 Amir Adva Acad, AS Mid-Atl Washington DC 1994 Andersen Robin CFI SWS Santa Rosa 2011 Anderson Anna Jo ATP NWS Wyoming 1982 Anderson Eileen B CFI SES Shreveport 1995 Anderson Katherine L CFI-Instrument SWS Orange County 2015 Andresen Diana LeSueur Instrument 2013 Fly Now SWS Tucson 1976 Anglin Mary Elaine CFI-Instrument Died 2020 NCS Michigan 2017 Anonsen Genevieve Multi-Engine SCS Pikes Peak 2007 Asmussen Lyndsay Ann ATP SWS Utah 2012 Assis Ruth Multi-Engine Israel Israel Jablonski- 1981 Bail Mary Suzanne Taylor Comm Multi-Engine SWS 1984 Bailey Martha "Janie" CFI SCS 1994 Bailey-Schmidt Susan P Bailey CFI SES Memphis 2010 Bair Wendy Type Rating, B737 NY/NJ 2011 Baker Ashley Comm Multi-Engine SWS Tucson 2003 Baker Jill Multi-Engine Instrument Also 2001 SWS Mission Bay 2018 Baker Julie Lyn Acad, Bach NWS Greater Seattle 1999 Baker Roberta F CFI Canada WCS 2010 Ballew Sue Seaplane SWS Santa Clara Valley 2010 Ballou Margaret H.L. CFI NCS All-Ohio 2019 Bandy Leslianne Instrument 2015 FlyNow SWS San Diego 1994 Barber Susan E Multi-Engine Mid-Atl W Washington 1989 Barker Linda CFI-Multi-Engine SWS Orange County 2005 Baron Janelle Nicole Instrument Also 2007 SCS Pikes Peak 2007 Baron Janelle Nicole Acad, Bach Also 2005. -

Saint Joseph County, Indiana Naturalization Record Index
Saint Joseph County, Indiana Naturalization Record Index 1st 1st Final Final Index Index Papers Papers Papers Papers Surname Given Accession Page Page Volume Page Volume Koantz Marie Louis 90044 259 0 138 24 Koavis Vinoze 90046 264 272 7 0 Kobacinski Frank 90077 252 258 2 0 Kobar Michael 90077 525 4 6 0 Kober Joseph 90045 293 159 23 0 Kobernick Emelean 90044 275 195 36 279 42 Koberski Inatius 90077 254 669 2 0 Kobiec Antonette 90044 290 154 42 0 Kobilski Joseph 90046 255 39 8 0 Kobleinski Frank 90077 522 330 3 0 Koblick John 90044 255 373 32 0 Koblick John J. 90044 303 162 40 0 Kobor Ignacz 90046 254 0 0 Kobor John 90077 399 435 4 0 Kobor Joseph Steve 90044 256 47 36 0 Kobor Louis 90044 258 276 37 0 Kobrin Paul 90045 333 273 25 0 Kobylaerk Andrew (Andrzy) 90044 249 156 34 266 32 Kobylarek Andizcj 90046 249 191 7 0 Kobylarek Andrew 90045 274 295 18 0 Kobzinski John Joseph 90044 303 0 61 45 Kocan Blaz 90046 250 0 0 Koch Charles 90077 251 317 4 0 Koch Edward 90077 252 394 1 0 Koch John 90077 396 0 31 11 Koch John 90045 275 498 19 228 16 Kochanouski Peter 90077 261 512 3 0 Kochanowski Frank 90046 252 329 7 0 Kochanowski Frank 90045 253 478 19 230 21 Kochanowski John 90045 304 280 27 0 Kochanowski Peter 90045 161 170 16 0 Kochanowski Wladyslaw 90045 265 308 15 0 Kochech John 90077 394 479 6 0 Kochendoerfer Antone 90077 249 118 2 0 Kochis Dan 90045 251 0 93 5 Kochler Matthies 90077 259 156 3 0 Kochler Valentine 90077 164 511 2 0 Kochneff Alex 90063 249 0 448 50 Kochod Frank 90077 522 708 4 23 7 Kocics Frank 90046 252 0 0 Kocics Frank 90045 252 62 12 0 Kockys Tony N. -

2020 Annual Report
Annual Report On Behalf of the Board of Trustees of New York Institute of Technology, thank you for helping us provide our students with the best education possible. To invest in higher education is to invest in our shared future—one that will be shaped (and reshaped) by the tech-savvy college students that you’ll find in our classrooms, incubators, maker spaces, and laboratories. Over the past year, we’ve tried to mitigate additional barriers to entry with our test-optional admissions policies, extended deadlines, and generous financial aid packages, but we still need to find new ways to deliver on our mission to offer access to opportunity. Your generosity continues to fuel our students’ learning experiences and professional development, helping them find solutions for the challenges of the 21st century, including those that have impacted them and all of us since March 2020. By making, doing, and innovating, they are discovering solutions that we, and the world at large, will benefit from. We are proud to fill an important niche in higher education, one that provides a tremendous opportunity to those who may think they cannot afford a college education. Our faculty serve as inspiring mentors who share their expertise while encouraging students to aspire to do great things. Our ever-increasing technology-infused, innovative, industry-grade resources and tools ensure that our students are “work ready” even before they graduate. Thanks to your support, the resources and unique classroom and cocurricular research and entrepreneurial experiences that we are able to offer our students will afford them a competitive edge as they jump-start their careers immediately upon graduation and sustain them throughout their careers. -

2020 Drayton Hall Donor List
PHILANTHROPY AT DRAYTON HALL Fiscal Year 2020 DRAYTON HALL PRESERVATION TRUST DONOR LISTING JULY 1, 2019 - JUNE 30, 2020 Our Mission Drayton Hall Preservation Trust fosters a deeper understanding of colonial America and the evolution of life in the South by discovering, researching, conserving, and interpreting the history, context, and culture of Drayton Hall. Contact: Halley Cella Erickson Director of Philanthropy Drayton Hall Preservation Trust | 3380 Ashley River Road Charleston, SC 29414 Phone: 252.617.0780 | Email: [email protected] Board of Trustees July 1, 2019 - June 30, 2020 Richard Almeida Chair W. Hampton Morris Vice-Chair Marilynn W. Hill Vice-Chair H. Montague Osteen, Jr. Treasurer Thomas W. Rash Secretary Nathan Berry Mary R. Black William B. Bodine Catherine Brown Braxton Frank W. Brumley John Cahill Amelia Cathcart Matthew Cochrane-Logan Steven Dopp Frank B. Drayton, Jr. Shannon C. Eaves Benjamin P. Jenkins, III Rise Johnson Cynthia Kellogg Kristopher B. King Douglas B. Lee Benjamin F. Lenhardt, Jr. Fulton D. Lewis Jr. Katherine Malone-France Margaret B. Pritchard Michael B. Prevost, Trustee Emeritus Henry B. Smythe, Jr. Anthony Wood 1 0 Cantey Technology Consulting Charleston Mercury Deep Eddy Vodka Duvall Events Edmunds Oast Brewery EventWorks Francis Marion Hotel Gilchrist/Bissell Wealth Management of Raymond James Glenn Keyes Architects Huss, Inc. Keystone Carpentry Co. LLC Koozer Painting Inc. Kuhn & Kuhn Law Firm LFA Architecture, LLC Lowcountry Audio Visual, LLC MIX Bartending Service PrintSouth Printing, Inc. Propel HR SMHa, Inc. The Middleton Group Totus Imaging Westbrook Brewing Company William Means Real Estate, LLC For more information on how your company can support Drayton Hall's mission through a sponsorship, please Corporate Friends of Drayton Hall 2 contact Caroline Schultz at [email protected] or 0 (843) 769-2611.