Reconciling Migration Models to the Americas with the Variation of North American Native Mitogenomes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Final Complete Dissertation Kua 1
Trends and Ontology of Artistic Practices of the Dorset Culture 800 BC - 1300 AD Hardenberg, Mari Publication date: 2013 Document version Early version, also known as pre-print Citation for published version (APA): Hardenberg, M. (2013). Trends and Ontology of Artistic Practices of the Dorset Culture 800 BC - 1300 AD. København: Det Humanistiske Fakultet, Københavns Universitet. Download date: 08. Apr. 2020 Trends and Ontology of Artistic Practices of the Dorset Culture 800 BC – 1300 AD Volume 1 By © Mari Hardenberg A Dissertation Submitted to the Ph.d.- School In Partial fulfillment of the Requirements for the Degree of Doctor of Philosophy SAXO-Institute, Department of Prehistoric Archaeology, Faculty of Humanities University of Copenhagen August 2013 Copenhagen Denmark ABSTRACT This dissertation examines the various artistic carvings produced by the hunter-gatherer Dorset people who occupied the eastern Arctic and temperate regions of Canada and Greenland between circa BC 800 – AD 1300. It includes considerations on how the carved objects affected and played a role in Dorset social life. To consider the role of people, things and other beings that may be said to play as actors in interdependent entanglements of actions, the agency/actor- network theory is employed. From this theoretical review an interpretation of social life as created by the ways people interact with the material world is presented. This framework is employed as a lens into the social role and meaning the carvings played in the Dorset society. The examined assemblages were recovered from a series of Dorset settlement sites, mainly in house, midden, and burial contexts, providing a substantive case study through which variations and themes of carvings are studied. -

Early Colonisation of Disko
- The Arctic Region of Disko - Early Colonisation of Disko by Peter Chapman Mountain Environment Early Colonisation of Disko The Arctic Region of Disko he first people to venture into the arctic were the Palaeo-Eskimo. Their movement into the arctic, which originated from the Bering Strait area between Siberia and Alaska, resulted in tact with them T perfecting methods for hunting marine animals throughout the year in the arctic conditions. This group of early Palaeo-Eskimos are known internationally as the Arctic Small Tool tradition (ASTt). Common to them are the small stone-tipped implements they used to survive. These Stone Age people spread along the northern coast of Alaska and Canada to Greenland in less than 100 years - an amazing speed considering the few numbers of people and the enormous distances involved. Their settlements where located close to their hunting grounds, either right on the coast by the sea ice, or along inlets from where they hunted land mammals such as reindeer (caribou) and muskoxen. Today, traces of these settlements are found on fossil terraces a little inland and often 30 to 40 metres above sea level due to continuing post glacial uplift of the land and changes in sea level since the time of inhabitation. The archaeologist Robert McGhee wrote that these people migrated into “the coldest, darkest and most barren regions ever inhabited by man”. Indeed, they were very bold to do so both in terms of coping with the harshness of the climate but also because of the psychological nature of their endeavours. The early Palaeo-Eskimo people who populated the arctic archipelago of Canada are called the Pre-Dorset Culture and two cultures populated areas of Greenland’s coast for the first time around 2400 BC. -

Genomic Study of the Ket: a Paleo-Eskimo-Related Ethnic Group with Significant Ancient North Eurasian Ancestry
Genomic study of the Ket: a Paleo-Eskimo-related ethnic group with significant ancient North Eurasian ancestry Pavel Flegontov1,2,3*, Piya Changmai1,§, Anastassiya Zidkova1,§, Maria D. Logacheva2,4, Olga Flegontova3, Mikhail S. Gelfand2,4, Evgeny S. Gerasimov2,4, Ekaterina E. Khrameeva5,2, Olga P. Konovalova4, Tatiana Neretina4, Yuri V. Nikolsky6,11, George Starostin7,8, Vita V. Stepanova5,2, Igor V. Travinsky#, Martin Tříska9, Petr Tříska10, Tatiana V. Tatarinova2,9,12* 1 Department of Biology and Ecology, Faculty of Science, University of Ostrava, Ostrava, Czech Republic 2 A.A.Kharkevich Institute for Information Transmission Problems, Russian Academy of Sciences, Moscow, Russian Federation 3 Institute of Parasitology, Biology Centre, Czech Academy of Sciences, České Budĕjovice, Czech Republic 4 Department of Bioengineering and Bioinformatics, Lomonosov Moscow State University, Moscow, Russian Federation 5 Skolkovo Institute of Science and Technology, Skolkovo, Russian Federation 6 Biomedical Cluster, Skolkovo Foundation, Skolkovo, Russian Federation 7 Russian State University for the Humanities, Moscow, Russian Federation 8 Russian Presidential Academy (RANEPA), Moscow, Russian Federation 9 Children's Hospital Los Angeles, Los Angeles, CA, USA 10 Instituto de Patologia e Imunologia Molecular da Universidade do Porto (IPATIMUP), Porto, Portugal 11 George Mason University, Fairfax, VA, USA 12 Spatial Science Institute, University of Southern California, Los Angeles, CA, USA *corresponding authors: P.F., email [email protected]; T.V.T., email [email protected] § the authors contributed equally # retired, former affiliation: Central Siberian National Nature Reserve, Bor, Krasnoyarsk Krai, Russian Federation. Abstract The Kets, an ethnic group in the Yenisei River basin, Russia, are considered the last nomadic hunter-gatherers of Siberia, and Ket language has no transparent affiliation with any language family. -

The Early Palaeoeskimo Period in the Eastern Arctic and the Labrador Early Pre-Dorset Period: a Reassessment
MASTER OF ARTS McMaster University (ANTHROPOLOGY) Hamilton, Ontario TITLE: The Early Palaeoeskimo Period in the Eastern Arctic and the Labrador Early Pre-Dorset period: a reassessment. AUTHOR: Karen Ryan SUPERVISOR: Dr. Peter Ramsden NUMBER OF PAGES: 121 II McMf,STER U['\iVEHSITY LIBRARY ABSTRACT By the end of the 1960s research in the Eastern North American Arctic had defined a single widespread Early Palaeoeskimo culture, dubbed Pre-Dorset since it preceded Late Palaeoeskimo Dorset culture. Subsequent investigations in Greenland resulted in the recognition of two other occupations, Independence I and Saqqaq, that, while different, were considered part of the Pre-Dorset manifestation. However, in the mid-1970s it was proposed that Independence I and Pre-Dorset should be considered culturally and temporally distinct. This classification system clearly divided the period and did not allow for interactions between the groups. While this proposal was initially questioned, it has come to dominate interpretation of the Early Palaeoeskimo period. At the same time as this framework was being promoted, a small number of Early Pre-Dorset sites were excavated in Labrador. Classified as Pre-Dorset, these sites nonetheless exhibited Independence I and Saqqaq influences. The reasons for this could not be fully explained, though a relationship between Labrador and the High Arctic was proposed. This thesis reevaluates the place of Labrador Early Pre-Dorset within the sphere of the Eastern Arctic following upon almost thirty years of archaeological work, both in Labrador and elsewhere in the Eastern Arctic. Recent evidence suggests that researchers must rethink their view of the Early Palaeoeskimo period and the vision of Independence I and Saqqaq relations. -

Abrupt Holocene Climate Change As an Important Factor for Human Migration in West Greenland
Abrupt Holocene climate change as an important factor for human migration in West Greenland William J. D’Andreaa,1,2, Yongsong Huanga,2, Sherilyn C. Fritzb, and N. John Andersonc aDepartment of Geological Sciences, Brown University, Providence, RI 02912; bDepartment of Earth and Atmospheric Sciences and School of Biological Sciences, University of Nebraska, Lincoln, NE 68588; cDepartment of Geography, Loughborough University, Leics LE11 3TU, United Kingdom Edited by Julian Sachs, University of Washington, Seattle, WA, and accepted by the Editorial Board April 22, 2011 (received for review January 31, 2011) West Greenland has had multiple episodes of human colonization to assess the influence of climate on observed patterns of human and cultural transitions over the past 4,500 y. However, the migration. explanations for these large-scale human migrations are varied, including climatic factors, resistance to adaptation, economic mar- An Alkenone-Based Paleotemperature Record for West ginalization, mercantile exploration, and hostile neighborhood Greenland interactions. Evaluating the potential role of climate change is com- Here we present a record of temperature variability with decadal- scale resolution for approximately the past 5,600 y from two plicated by the lack of quantitative paleoclimate reconstructions 14 near settlement areas and by the relative stability of Holocene independently C dated, finely laminated sediment cores from two meromictic lakes, Braya Sø and Lake E (approximately temperature derived from ice cores atop the Greenland ice sheet. ′ Here we present high-resolution records of temperature over the 10 km apart) in Kangerlussuaq, West Greenland (67° 0 N, 50° 42′ W; Fig. 1 and SI Appendix, Section 1). -
Executive Summary
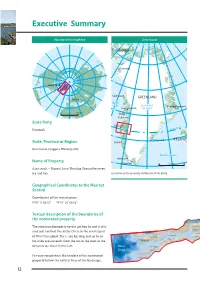
Nipisat_MANDAG.qxp_Aasivissuit 13/12/16 13:05 Page 12 Executive Summary Northern hemisphere Greenland 80°W 70°W 60°W 50°W 40°W 30°W 20°W 10°W 0°W CANADA Qaanaaq 75°N Arctic Circle Greenland Upernavik GREENLAND Denmark Baffin Bay Greenland Ittoqqortoormiit 70°N Uummannaq Ice sheet 0 500 km Disko Ilulissat State Party Sisimiut Circle Denmark Arctic 65°N Ammassalik ICELAND State, Province or Region Nuuk Greenland, Qeqqata Municipality Atlantic Ocean Ivittuut Name of Property 60°N Qaqortoq 500 km Aasivissuit – Nipisat. Inuit Hunting Ground between Ice and Sea Location of the property within the State Party. Geographical Coordinates to the Nearest Second Coordinates of the central point: N 67° 3' 50.15" W 51° 25' 59.54" Textual description of the boundaries of the nominated property TASEQ QAT SAQ QAA TASERSUAQ The nominated property covers 417,800 ha and is situ- MALIGISSAP QAAVA Niaqornarsuaq Qarlissuit ated just north of the Arctic Circle in the central part O Q E R L M Akulleq A SARFANNGUIT Akuliaruseq of West Greenland. The c. 235 km long and up to 20 SARFANNGUAQ SAQQARLIIT Iter las su a Nipisat I K E R T O O Q q km wide area extends from the sea in the west to the C dynamic ice sheet in the east. Davis IKERASAARSUK Strait SALLERSUAQ For easy recognition, the borders of the nominated SAQQAQ property follow the natural lines of the landscape, 12 Nipisat_MANDAG.qxp_Aasivissuit 13/12/16 13:05 Page 13 Aasivissuit – Nipisat Inuit Hunting Ground between Ice and Sea Nominated area Greenland Ice sheet 67°N Kangerlussuaq Sisimiut 67°N Sarfannguit Davis Strait Itilleq 0 10 20 30 km 66°N 54°W 52°W 50°W 66°N Location of the property within the region. -

Paleoeskimo Dogs of the Eastern Arctic DARCY F
ARCTIC VOL. 55, NO. 1 (MARCH 2002) P. 44–56 Paleoeskimo Dogs of the Eastern Arctic DARCY F. MOREY1 and KIM AARIS-SØRENSEN2 (Received 28 June 2000; accepted in revised form 11 June 2001) ABSTRACT. Sled or pack dogs have been perceived as an integral part of traditional life in the Eastern Arctic. This perception stems from our knowledge of the lifeway of recent Thule and modern Inuit peoples, among whom dog sledding has often been an important means of transportation. In contrast, the archaeological record of preceding Paleoeskimo peoples indicates that dogs were sparse at most, and probably locally absent for substantial periods. This pattern is real, not an artifact of taphonomic biases or difficulties in distinguishing dog from wolf remains. Analysis of securely documented dog remains from Paleoeskimo sites in Greenland and Canada underscores the sporadic presence of only small numbers of dogs, at least some of which were eaten. This pattern should be expected. Dogs did not, and could not, assume a conspicuous role in North American Arctic human ecology outside the context of several key features of technology and subsistence production associated with Thule peoples. Key words: Thule, Dorset, Inuit, zooarchaeology, dog, sled, transportation, economics, Greenland, eastern Arctic RÉSUMÉ. On a toujours considéré les chiens de traîneau ou chiens de somme comme faisant partie intégrante de la vie traditionnelle dans l’Arctique oriental. Cette perception vient de nos connaissances sur le mode de vie des derniers Thulés et des Inuits modernes, chez qui le traîneau à chiens a souvent représenté un mode de transport majeur. En revanche, les données archéologiques des peuples paléoesquimaux qui ont précédé Thulés et Inuits révèlent que les chiens étaient tout au plus clairsemés, et probablement absents localement pendant de longues périodes. -

The Arctic Small Tool Tradition Fifty Years On
THE ARCTIC SMALL TOOL TRADITION FIFTY YEARS ON Daniel Odess University of Alaska Museum, 907 Yukon Drive / Box 756960, Fairbanks, Alaska 99775. [email protected] Abstract: The Arctic Small Tool tradition (ASTt) encompasses several culture complexes in Alaska, Canada, and Greenland. Research on the Alaskan members of the tradition has not kept pace with that in the rest of the North American Arctic. Despite the passage of more than fifty years since its discovery, there is still a great deal we do not know about the Denbigh Flint Complex, and much of what we think we know is based on received wisdom and ethnographic analogy rather than direct archaeological evidence. This paper assesses the state of our knowledge about the ASTt in Alaska and situates it within the broader framework of Arctic prehistory. Keywords: Alaska Archaeology, Arctic Prehistory, Middle Holocene, Denbigh Flint Complex Nearly fifty years ago, a young William Irving re- ognized and our ability to do so realized (Reimer et al. flected on the similarities between the small, delicately 2004; Stuiver et al. 1998). Accelerator mass spectrom- flaked stone tools that had recently been discovered in etry (AMS) has been developed and now permits us to Alaska (Giddings 1949, 1951), Canada (Giddings 1956; date minute samples of organic matter from sites that Harp 1958), and Greenland (Knuth 1954; Larsen and would have been undateable in 1980. Equally important, Meldgaard 1958; Meldgaard 1952), and suggested that AMS permits us to choose samples for dating based on they shared a common historical origin. Aware of the the most appropriate context and association rather than need for consistency in archaeological systematics and on the basis of sample size. -

Greenland – an Arctic Society
By Svend Kolte, director of the Greenlanders’ House in Århus, eskimologist, MA. The foreign student’s guide to: Greenland – an Arctic Society 1. The Geography and Climate of Greenland Greenland is an island situated in the North Atlantic Ocean, east of the North American continent. The northern tip of Greenland, Cape Morris Jesup, lies only 740 km south of the North Pole, thus being the northernmost land area in the world. The distance from this point to the southern tip of the island, Cape Farewell, is 2,670 km. The maximum distance from the East to the West coast is 1,050 km. Greenland is the largest island in the world. With 2,166,086 sq. km. it is much larger than e.g. New Guinea or Madagascar. And almost as large as Spain, Italy, France, United Kingdom, Germany and Poland together! However, most of the island is covered with ice, the so-called Ice Cap of Greenland, which covers about 81% of the total area. The Ice Cap is the largest body of ice in the northern hemisphere, comparable only to the Ice Cap of the South Pole. The ice-free rim of land, between the Ice Cap and the sea, is very narrow in places, sometimes broader, and comprises about 410,000 sq. km. – comparable to the area of Sweden. The climate of the country is everywhere arctic, i.e. nowhere does the average summer temperature of July reach 10o. Therefore no extensive forests are found, only clusters of small trees in the very south of the island. -

A Saqqaq Culture Site in Sisimiut, Central West Greenland, 2004 Eisbn 978-87-635-2626-5, Monographs on Greenland | Meddelelser Om Grønland, Vol
Museum Tusculanum Press - University of Copenhagen :: www.mtp.dk :: [email protected] Published in cooperation with Sisimiut Museum and SILA, the Greenland Research Centre at the National Museum of Denmark. SISIMIUT KATERSUGAASIVIAT SISIMIUT MUSEUM Anne Birgitte Gotfredsen & Tinna Møbjerg: Nipisat - a Saqqaq culture site in Sisimiut, central West Greenland, 2004 eISBN 978-87-635-2626-5, Monographs on Greenland | Meddelelser om Grønland, vol. 331 (ISSN 0025-6676) Man and Society, vol. 31 (ISSN 0106-1062), http://www.mtp.hum.ku.dk/details.asp?eln=201396 Museum Tusculanum Press - University of Copenhagen :: www.mtp.dk :: [email protected] Nipisat – a Saqqaq Culture Site in Sisimiut, Central West Greenland Anne Birgitte Gotfredsen and Tinna Møbjerg – with a contribution by Kaj Strand Petersen and Ella Hoch Meddelelser om Grønland · Man & Society 31 Anne Birgitte Gotfredsen & Tinna Møbjerg: Nipisat - a Saqqaq culture site in Sisimiut, central West Greenland, 2004 eISBN 978-87-635-2626-5, Monographs on Greenland | Meddelelser om Grønland, vol. 331 (ISSN 0025-6676) Man and Society, vol. 31 (ISSN 0106-1062), http://www.mtp.hum.ku.dk/details.asp?eln=201396 Gotfredsen, A.B. and Møbjerg, T. Nipisat - a Saqqaq Culture Site in Sisimiut, Central West Greenland. Monographs on Greenland | Meddelelser om Grønland, vol. 331 (ISSN 0025-6676) Man and Society, vol. 31 (ISSN 0106-1062) © 2004 by SILA and Danish Polar Center (ISBN 87-90369-73-4) MuseumISBN 978-87-635-1264-0 Tusculanum Press (Museum - University Tusculanum of Copenhagen Press) :: www.mtp.dk :: [email protected] eISBN 978-87-635-2626-5 (Museum Tusculanum Press: unchanged PDF-version of print edition) Publishing editor Kirsten Caning Printed by Special-Trykkeriet Viborg a-s No part of this publication may be reproduced in any form without the written permission of the copyright owners. -

Identifying Pre-Dorset Structural Features on Southern Baffin Island
Document generated on 09/25/2021 10:51 p.m. Études/Inuit/Studies Identifying Pre-Dorset structural features on southern Baffin Island: Challenges and considerations for alternative sampling methods Identification de structures prédorsétiennes en Terre de Baffin méridionale: méthodes alternatives d'échantillonnage S. Brooke Milne Architecture paléoesquimaude Article abstract Palaeoeskimo Architecture This paper describes an isolated Pre-Dorset component found at the Volume 27, Number 1-2, 2003 Tungatsivvik (KkDo-3) site on southern Baffin Island. Micro-debitage analysis and systematic core sampling are proposed as a combined strategy to facilitate URI: https://id.erudit.org/iderudit/010796ar future investigations of Pre-Dorset occupations in this region. DOI: https://doi.org/10.7202/010796ar See table of contents Publisher(s) Association Inuksiutiit Katimajiit Inc. ISSN 0701-1008 (print) 1708-5268 (digital) Explore this journal Cite this article Milne, S. B. (2003). Identifying Pre-Dorset structural features on southern Baffin Island: Challenges and considerations for alternative sampling methods. Études/Inuit/Studies, 27(1-2), 67–90. https://doi.org/10.7202/010796ar Tous droits réservés © La revue Études/Inuit/Studies, 2003 This document is protected by copyright law. Use of the services of Érudit (including reproduction) is subject to its terms and conditions, which can be viewed online. https://apropos.erudit.org/en/users/policy-on-use/ This article is disseminated and preserved by Érudit. Érudit is a non-profit inter-university consortium of the Université de Montréal, Université Laval, and the Université du Québec à Montréal. Its mission is to promote and disseminate research. https://www.erudit.org/en/ Identifying Pre-Dorset structural features on southern Baffin Island: Challenges and considerations for alternative sampling methods S. -

Thule-Culture (Inland) Tasersiaq – Aussivissuit the L 151, Napasorsuaq, Is from a Human Bone and Too Old
Culture historical significance on areas Tasersiaq and Tarsartuup Tasersua in West Greenland & Suggestions for Salvage Archaeology and Documentation in Case of Damming Lakes - Report prepared for ALCOA, - May 2009. By Pauline K. Knudsen with contributions from Claus Andreasen Nunatta Katersugaasivia Allagaateqarfialu Greenland National Museum and Archives May 2009 1 Table of Contents Introduction……………………………………………………………………………….. 3 The Culture historical background………………………………………………………... 4 Results from the surveys………………………………………………………………….. 5 Finds by Lakes in Nuuk area, 6g ..……………………………………………………… . 6 Finds by Tasersiaq, 7e……………………………………………………………………. 9 The Culture Historical Significance of Finds……………………………………………... 13 Suggestion for Preservation of Tasersiaq…………………………………………………. 15 Suggestion for Salvage Archaeology and documentation in Case of Damming of Lakes……………………………………………. ………………………………………… 17 Proposal for Research Questions………………………………………………………….. 18 Summary of Cultural Remains to be Excavated…...……………………………………… 19 Description of Settlements and Cultural remains proposed for excavation…..…….…… 20 Tarsartuup Tasersua and adjacent areas (6g)……………………………………………... 20 Tasersiaq (7e)……………………………………………………………………………… 26 List of References………………………………………………………………………… 34 Appendix A Functional Definitions of types of Cultural Remains…………………………………….. 36 Appendix B Summary of Cultural Remains for Excavation …………………………………………… 39 Appendix C Radiocarbon dates..……………………………………………………………………….. 41 2 Introduction Archaeological studies were performed by