Accurate Contact-Based Modelling of Repeat Proteins Predicts the Structure Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Ankrd9 Is a Metabolically-Controlled Regulator of Impdh2 Abundance and Macro-Assembly
ANKRD9 IS A METABOLICALLY-CONTROLLED REGULATOR OF IMPDH2 ABUNDANCE AND MACRO-ASSEMBLY by Dawn Hayward A dissertation submitted to The Johns Hopkins University in conformity with the requirements of the degree of Doctor of Philosophy Baltimore, Maryland April 2019 ABSTRACT Members of a large family of Ankyrin Repeat Domains proteins (ANKRD) regulate numerous cellular processes by binding and changing properties of specific protein targets. We show that interactions with a target protein and the functional outcomes can be markedly altered by cells’ metabolic state. ANKRD9 facilitates degradation of inosine monophosphate dehydrogenase 2 (IMPDH2), the rate-limiting enzyme in GTP biosynthesis. Under basal conditions ANKRD9 is largely segregated from the cytosolic IMPDH2 by binding to vesicles. Upon nutrient limitation, ANKRD9 loses association with vesicles and assembles with IMPDH2 into rod-like structures, in which IMPDH2 is stable. Inhibition of IMPDH2 with Ribavirin favors ANKRD9 binding to rods. The IMPDH2/ANKRD9 assembly is reversed by guanosine, which restores association of ANKRD9 with vesicles. The conserved Cys109Cys110 motif in ANKRD9 is required for the vesicles-to-rods transition as well as binding and regulation of IMPDH2. ANKRD9 knockdown increases IMPDH2 levels and prevents formation of IMPDH2 rods upon nutrient limitation. Thus, the status of guanosine pools affects the mode of ANKRD9 action towards IMPDH2. Advisor: Dr. Svetlana Lutsenko, Department of Physiology, Johns Hopkins University School of Medicine Second reader: -

Repeat Proteins Challenge the Concept of Structural Domains
844 Biochemical Society Transactions (2015) Volume 43, part 5 Repeat proteins challenge the concept of structural domains Rocıo´ Espada*1, R. Gonzalo Parra*1, Manfred J. Sippl†, Thierry Mora‡, Aleksandra M. Walczak§ and Diego U. Ferreiro*2 *Protein Physiology Lab, Dep de Qu´ımica Biologica, ´ Facultad de Ciencias Exactas y Naturales, UBA-CONICET-IQUIBICEN, Buenos Aires, C1430EGA, Argentina †Center of Applied Molecular Engineering, Division of Bioinformatics, Department of Molecular Biology, University of Salzburg, 5020 Salzburg, Austria ‡Laboratoire de physique statistique, CNRS, UPMC and Ecole normale superieure, ´ 24 rue Lhomond, 75005 Paris, France §Laboratoire de physique theorique, ´ CNRS, UPMC and Ecole normale superieure, ´ 24 rue Lhomond, 75005 Paris, France Abstract Structural domains are believed to be modules within proteins that can fold and function independently. Some proteins show tandem repetitions of apparent modular structure that do not fold independently, but rather co-operate in stabilizing structural forms that comprise several repeat-units. For many natural repeat-proteins, it has been shown that weak energetic links between repeats lead to the breakdown of co-operativity and the appearance of folding sub-domains within an apparently regular repeat array. The quasi-1D architecture of repeat-proteins is crucial in detailing how the local energetic balances can modulate the folding dynamics of these proteins, which can be related to the physiological behaviour of these ubiquitous biological systems. Introduction and between repeats, challenging the concept of structural It was early on noted that many natural proteins typically domain. collapse stretches of amino acid chains into compact units, defining structural domains [1]. These domains typically correlate with biological activities and many modern proteins can be described as composed by novel ‘domain arrange- Definition of the repeat-units ments’ [2]. -

A Structural Guide to Proteins of the NF-Kb Signaling Module
Downloaded from http://cshperspectives.cshlp.org/ on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press A Structural Guide to Proteins of the NF-kB Signaling Module Tom Huxford1 and Gourisankar Ghosh2 1Department of Chemistry and Biochemistry, San Diego State University, 5500 Campanile Drive, San Diego, California 92182-1030 2Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, California 92093-0375 Correspondence: [email protected] The prosurvival transcription factor NF-kB specifically binds promoter DNA to activate target gene expression. NF-kB is regulated through interactions with IkB inhibitor proteins. Active proteolysis of these IkB proteins is, in turn, under the control of the IkB kinase complex (IKK). Together, these three molecules form the NF-kB signaling module. Studies aimed at charac- terizing the molecular mechanisms of NF-kB, IkB, and IKK in terms of their three-dimen- sional structures have lead to a greater understanding of this vital transcription factor system. F-kB is a master transcription factor that from the perspective of their three-dimensional Nresponds to diverse cell stimuli by activat- structures. ing the expression of stress response genes. Multiple signals, including cytokines, growth factors, engagement of the T-cell receptor, and NF-kB bacterial and viral products, induce NF-kB Introduction to NF-kB transcriptional activity (Hayden and Ghosh 2008). A point of convergence for the myriad NF-kB was discovered in the laboratory of of NF-kB inducing signals is the IkB kinase David Baltimore as a nuclear activity with bind- complex (IKK). Active IKK in turn controls ing specificity toward a ten-base-pair DNA transcription factor NF-kB by regulating pro- sequence 50-GGGACTTTCC-30 present within teolysis of the IkB inhibitor protein (Fig. -
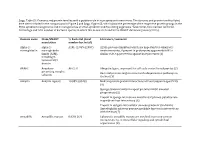
Supp. Table S2: Domains and Protein Families with a Putative Role in Host-Symbiont Interactions
Supp. Table S2: Domains and protein families with a putative role in host-symbiont interactions. The domains and protein families listed here were included in the comparisons in Figure 5 and Supp. Figure S5, which show the percentage of the respective protein groups in the Riftia symbiont metagenome and in metagenomes of other symbiotic and free-living organisms. % bacterial, total number bacterial: Percentage and total number of bacterial species in which this domain is found in the SMART database (January 2019). Domain name Pfam/SMART % bacterial (total Literature/comment annotation number bacterial) Alpha-2- alpha-2- A2M: 42.05% (2057) A2Ms: protease inhibitors which are important for eukaryotic macroglobulin macroglobulin innate immunity, if present in prokaryotes apparently fulfill a family (A2M), similar role, e.g. protection against host proteases (1) including N- terminal MG1 domain ANAPC Anaphase- APC2: 0 Ubiquitin ligase, important for cell cycle control in eukaryotes (2) promoting complex Bacterial proteins might interact with ubiquitination pathways in subunits the host (3) Ankyrin Ankyrin repeats 10.88% (8348) Mediate protein-protein interactions without sequence specificity (4) Sponge symbiont ankyrin-repeat proteins inhibit amoebal phagocytosis (5) Present in sponge microbiome metatranscriptomes, putative role in symbiont-host interactions (6) Present in obligate intracellular amoeba symbiont Candidatus Amoebophilus asiaticus genome, probable function in interactions with the host (7) Armadillo Armadillo repeats 0.83% (67) -

Identification and Characterisation of Human Cytomegalovirus-Mediated Degradation of Helicase-Like Transcription Factor
Identification and Characterisation of Human Cytomegalovirus-Mediated Degradation of Helicase-Like Transcription Factor Kai-Min Lin Department of Medicine Cambridge Institute for Medical Research University of Cambridge This dissertation is submitted for the degree of Doctor of Philosophy Fitzwilliam College September 2020 Declaration I hereby declare, that except where specific reference is made to the work of others, the contents of this dissertation are original and have not been submitted in whole or in part for consideration for any other degree of qualification in this, or any other university. This dissertation is the result of my own work and includes nothing which is the outcome of work done in collaboration except as where specified in the text and acknowledgments. This dissertation does not exceed the specified word limit of 60,000 words as defined by the Degree Committee, excluding figures, photographs, tables, appendices and bibliography. Kai-Min Lin September, 2020 I Summary Identification and characterisation of human cytomegalovirus-mediated degradation of helicase-like transcription factor Kai-Min Lin Viruses are known to degrade host factors that are important in innate antiviral immunity in order to infect successfully. To systematically identify host proteins targeted for early degradation by human cytomegalovirus (HCMV), the lab developed orthogonal screens using high resolution multiplexed mass spectrometry. Taking advantage of broad and selective proteasome and lysosome inhibitors, proteasomal degradation was found to be heavily exploited by HCMV. Several known antiviral restriction factors, including components of cellular promyelocytic leukemia (PML) were enriched in a shortlist of proteasomally degraded proteins during infection. A particularly robust novel ‘hit’ was helicase-like transcription factor (HLTF), a DNA repair protein that participates in error-free repair of stalled replication forks. -

Appendix 2. Significantly Differentially Regulated Genes in Term Compared with Second Trimester Amniotic Fluid Supernatant
Appendix 2. Significantly Differentially Regulated Genes in Term Compared With Second Trimester Amniotic Fluid Supernatant Fold Change in term vs second trimester Amniotic Affymetrix Duplicate Fluid Probe ID probes Symbol Entrez Gene Name 1019.9 217059_at D MUC7 mucin 7, secreted 424.5 211735_x_at D SFTPC surfactant protein C 416.2 206835_at STATH statherin 363.4 214387_x_at D SFTPC surfactant protein C 295.5 205982_x_at D SFTPC surfactant protein C 288.7 1553454_at RPTN repetin solute carrier family 34 (sodium 251.3 204124_at SLC34A2 phosphate), member 2 238.9 206786_at HTN3 histatin 3 161.5 220191_at GKN1 gastrokine 1 152.7 223678_s_at D SFTPA2 surfactant protein A2 130.9 207430_s_at D MSMB microseminoprotein, beta- 99.0 214199_at SFTPD surfactant protein D major histocompatibility complex, class II, 96.5 210982_s_at D HLA-DRA DR alpha 96.5 221133_s_at D CLDN18 claudin 18 94.4 238222_at GKN2 gastrokine 2 93.7 1557961_s_at D LOC100127983 uncharacterized LOC100127983 93.1 229584_at LRRK2 leucine-rich repeat kinase 2 HOXD cluster antisense RNA 1 (non- 88.6 242042_s_at D HOXD-AS1 protein coding) 86.0 205569_at LAMP3 lysosomal-associated membrane protein 3 85.4 232698_at BPIFB2 BPI fold containing family B, member 2 84.4 205979_at SCGB2A1 secretoglobin, family 2A, member 1 84.3 230469_at RTKN2 rhotekin 2 82.2 204130_at HSD11B2 hydroxysteroid (11-beta) dehydrogenase 2 81.9 222242_s_at KLK5 kallikrein-related peptidase 5 77.0 237281_at AKAP14 A kinase (PRKA) anchor protein 14 76.7 1553602_at MUCL1 mucin-like 1 76.3 216359_at D MUC7 mucin 7, -

A New Census of Protein Tandem Repeats and Their Relationship with Intrinsic Disorder
G C A T T A C G G C A T genes Article A New Census of Protein Tandem Repeats and Their Relationship with Intrinsic Disorder Matteo Delucchi 1,2 , Elke Schaper 1,2,† , Oxana Sachenkova 3,‡, Arne Elofsson 3 and Maria Anisimova 1,2,* 1 ZHAW Life Sciences und Facility Management, Applied Computational Genomics, 8820 Wädenswil, Switzerland; [email protected] 2 Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland 3 Science of Life Laboratory, Department of Biochemistry and Biophysics, Stockholm University, 106 91 Stockholm, Sweden * Correspondence: [email protected]; Tel.: +41-(0)58-934-5882 † Present address: Carbon Delta AG, 8002 Zürich, Switzerland. ‡ Present address: Vildly AB, 385 31 Kalmar, Sweden. Received: 9 March 2020; Accepted: 1 April 2020; Published: 9 April 2020 Abstract: Protein tandem repeats (TRs) are often associated with immunity-related functions and diseases. Since that last census of protein TRs in 1999, the number of curated proteins increased more than seven-fold and new TR prediction methods were published. TRs appear to be enriched with intrinsic disorder and vice versa. The significance and the biological reasons for this association are unknown. Here, we characterize protein TRs across all kingdoms of life and their overlap with intrinsic disorder in unprecedented detail. Using state-of-the-art prediction methods, we estimate that 50.9% of proteins contain at least one TR, often located at the sequence flanks. Positive linear correlation between the proportion of TRs and the protein length was observed universally, with Eukaryotes in general having more TRs, but when the difference in length is taken into account the difference is quite small. -

Intrinsic Disorder in Tetratricopeptide Repeat Proteins
International Journal of Molecular Sciences Article Intrinsic Disorder in Tetratricopeptide Repeat Proteins 1, 1, 1, 2 Nathan W. Van Bibber y, Cornelia Haerle y, Roy Khalife y, Bin Xue and Vladimir N. Uversky 1,3,4,* 1 Department of Molecular Medicine Morsani College of Medicine, University of South Florida, 12901 Bruce B. Downs Blvd., Tampa, FL 33612, USA; [email protected] (N.W.V.B.); [email protected] (C.H.); [email protected] (R.K.) 2 Department of Cell Biology, Microbiology and Molecular Biology, School of Natural Sciences and Mathematics, College of Arts and Sciences, University of South Florida, Tampa, FL 33620, USA; [email protected] 3 USF Health Byrd Alzheimer’s Research Institute, Morsani College of Medicine, University of South Florida, 12901 Bruce B. Downs Blvd., Tampa, FL 33612, USA 4 Institute for Biological Instrumentation, Russian Academy of Sciences, Federal Research Center “Pushchino Scientific Center for Biological Research of the Russian Academy of Sciences”, 4 Institutskaya St., Pushchino, 142290 Moscow Region, Russia * Correspondence: [email protected]; Tel.: +1-813-974-5816; Fax: +1-813-974-7357 These authors contributed equally to this work. y Received: 21 April 2020; Accepted: 22 May 2020; Published: 25 May 2020 Abstract: Among the realm of repeat containing proteins that commonly serve as “scaffolds” promoting protein-protein interactions, there is a family of proteins containing between 2 and 20 tetratricopeptide repeats (TPRs), which are functional motifs consisting of 34 amino acids. The most distinguishing feature of TPR domains is their ability to stack continuously one upon the other, with these stacked repeats being able to affect interaction with binding partners either sequentially or in combination. -

Repeatsdb in 2021: Improved Data And
RepeatsDB in 2021: improved data and extended classification for protein tandem repeat structures Lisanna Paladin, Martina Bevilacqua, Sara Errigo, Damiano Piovesan, Ivan Mičetić, Marco Necci, Alexander Miguel Monzon, Maria Laura Fabre, Jose luis Lopez, Juliet Nilsson, et al. To cite this version: Lisanna Paladin, Martina Bevilacqua, Sara Errigo, Damiano Piovesan, Ivan Mičetić, et al.. RepeatsDB in 2021: improved data and extended classification for protein tandem repeat structures. Nucleic Acids Research, Oxford University Press, 2020, 10.1093/nar/gkaa1097. hal-03089312 HAL Id: hal-03089312 https://hal.archives-ouvertes.fr/hal-03089312 Submitted on 4 Jan 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Page 1 of 11 Nucleic Acids Research 1 2 RepeatsDB in 2021: improved data and 3 4 5 extended classification for protein tandem 6 7 repeat structures 8 9 10 Lisanna Paladin1, Martina Bevilacqua1, Sara Errigo1, Damiano Piovesan1, Ivan Mičetić1, Marco Necci1, 11 Alexander Miguel Monzon1, Maria Laura Fabre2, Jose Luis Lopez2, Juliet F. Nilsson2, Javier Rios3, Pablo 12 3 3 3 4 13 Lorenzano Menna , Maia Cabrera , Martin Gonzalez Buitron , Mariane Gonçalves Kulik , Sebastian 14 Fernandez-Alberti3, Maria Silvina Fornasari3, Gustavo Parisi3, Antonio Lagares2, Layla Hirsh5, Miguel A. -

WSB1: from Homeostasis to Hypoxia Moinul Haque1,2,3, Joseph Keith Kendal1,2,3, Ryan Matthew Macisaac1,2,3 and Douglas James Demetrick1,2,3,4*
Haque et al. Journal of Biomedical Science (2016) 23:61 DOI 10.1186/s12929-016-0270-3 REVIEW Open Access WSB1: from homeostasis to hypoxia Moinul Haque1,2,3, Joseph Keith Kendal1,2,3, Ryan Matthew MacIsaac1,2,3 and Douglas James Demetrick1,2,3,4* Abstract The wsb1 gene has been identified to be important in developmental biology and cancer. A complex transcriptional regulation of wsb1 yields at least three functional transcripts. The major expressed isoform, WSB1 protein, is a substrate recognition protein within an E3 ubiquitin ligase, with the capability to bind diverse targets and mediate ubiquitinylation and proteolytic degradation. Recent data suggests a new role for WSB1 as a component of a neuroprotective pathway which results in modification and aggregation of neurotoxic proteins such as LRRK2 in Parkinson’s Disease, via an unusual mode of protein ubiquitinylation. WSB1 is also involved in thyroid hormone homeostasis, immune regulation and cellular metabolism, particularly glucose metabolism and hypoxia. In hypoxia, wsb1 is a HIF-1 target, and is a regulator of the degradation of diverse proteins associated with the cellular response to hypoxia, including HIPK2, RhoGDI2 and VHL. Major roles are to both protect HIF-1 function through degradation of VHL, and decrease apoptosis through degradation of HIPK2. These activities suggest a role for wsb1 in cancer cell proliferation and metastasis. As well, recent work has identified a role for WSB1 in glucose metabolism, and perhaps in mediating the Warburg effect in cancer cells by maintaining the function of HIF1. Furthermore, studies of cancer specimens have identified dysregulation of wsb1 associated with several types of cancer, suggesting a biologically relevant role in cancer development and/or progression. -

Intrinsic Disorder of the BAF Complex: Roles in Chromatin Remodeling and Disease Development
International Journal of Molecular Sciences Article Intrinsic Disorder of the BAF Complex: Roles in Chromatin Remodeling and Disease Development Nashwa El Hadidy 1 and Vladimir N. Uversky 1,2,* 1 Department of Molecular Medicine, Morsani College of Medicine, University of South Florida, 12901 Bruce B. Downs Blvd. MDC07, Tampa, FL 33612, USA; [email protected] 2 Laboratory of New Methods in Biology, Institute for Biological Instrumentation of the Russian Academy of Sciences, Federal Research Center “Pushchino Scientific Center for Biological Research of the Russian Academy of Sciences”, Pushchino, 142290 Moscow Region, Russia * Correspondence: [email protected]; Tel.: +1-813-974-5816; Fax: +1-813-974-7357 Received: 20 September 2019; Accepted: 21 October 2019; Published: 23 October 2019 Abstract: The two-meter-long DNA is compressed into chromatin in the nucleus of every cell, which serves as a significant barrier to transcription. Therefore, for processes such as replication and transcription to occur, the highly compacted chromatin must be relaxed, and the processes required for chromatin reorganization for the aim of replication or transcription are controlled by ATP-dependent nucleosome remodelers. One of the most highly studied remodelers of this kind is the BRG1- or BRM-associated factor complex (BAF complex, also known as SWItch/sucrose non-fermentable (SWI/SNF) complex), which is crucial for the regulation of gene expression and differentiation in eukaryotes. Chromatin remodeling complex BAF is characterized by a highly polymorphic structure, containing from four to 17 subunits encoded by 29 genes. The aim of this paper is to provide an overview of the role of BAF complex in chromatin remodeling and also to use literature mining and a set of computational and bioinformatics tools to analyze structural properties, intrinsic disorder predisposition, and functionalities of its subunits, along with the description of the relations of different BAF complex subunits to the pathogenesis of various human diseases. -

Structural and Biochemical Consequences of Disease-Causing Mutations in the Ankyrin Repeat Domain of the Human TRPV4 Channel
Structural and Biochemical Consequences of Disease-Causing Mutations in the Ankyrin Repeat Domain of the Human TRPV4 Channel The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Inada, Hitoshi, Erik Procko, Marcos Sotomayor, and Rachelle Gaudet. 2012. Structural and biochemical consequences of disease- causing mutations in the ankyrin repeat domain of the human TRPV4 channel. Biochemistry 51(31): 6195-6206. Published Version doi:10.1021/bi300279b Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:11688820 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Structural and biochemical consequences of disease-causing mutations in the ankyrin repeat domain of the human TRPV4 channel Hitoshi Inada†, Erik Procko†,‡, Marcos Sotomayor†,§, and Rachelle Gaudet†,* †Department of Molecular and Cellular Biology, Harvard University, 52 Oxford Street, Cambridge, MA 02138 ‡Present address: Howard Hughes Medical Institute and Department of Biochemistry, University of Washington, Seattle, WA 98195, USA §Howard Hughes Medical Institute and Department of Neurobiology, Harvard Medical School, Boston, MA 02115, USA AUTHOR EMAIL ADDRESS: [email protected] TITLE RUNNING HEAD: Structural analysis of human TRPV4-ARD CORRESPONDING AUTHOR FOOTNOTE: *To whom correspondence should be addressed: Rachelle Gaudet, Department of Molecular and Cellular Biology, Harvard University, 52 Oxford Street, Cambridge, MA 02138, USA, Tel.: (617) 495-5616; Fax: (617) 496-9684; E-mail: [email protected] FOOTNOTES: This work was funded by NIH grant R01GM081340 to RG.