PRILACTONE, INN-Spironolactone
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Dehydroepiandrosterone – Is the Fountain of Youth Drying Out?
Physiol. Res. 52: 397-407, 2003 MINIREVIEW Dehydroepiandrosterone – Is the Fountain of Youth Drying Out? P. CELEC 1,2, L. STÁRKA3 1Faculty of Medicine, 2Faculty of Natural Sciences, Comenius University, Bratislava, Slovakia and 3Institute of Endocrinology, Prague, Czech Republic Received September 15, 2002 Accepted October 7, 2002 Summary Dehydroepiandrosterone (DHEA) and its sulphate-bound form (DHEAS) are important steroids mainly of adrenal origin. Their physiological and pathophysiological functions are not yet fully identified, although a number of various possible features have been hypothesized. Most popular is the description of the “hormone of youth” as the long-term dynamics of DHEA levels are characterized by a sharp age-related decline in the late adulthood and later. Low levels of DHEA are, however, associated not only with the ageing process but also with diabetes mellitus, cardiovascular diseases and some neurological or immunological entities. In the past decade, a number of brief studies have concentrated on these relationships and also on the role of exogenous DHEA in health, disease and human well-being. This article tries to summarize some of the most important facts achieved recently. Key words Dehydroepiandrosterone • Intracrinology • Hormone replacement therapy • Steroids Introduction functions: 1) DHEA is an endogenous metabolite that cannot be patented so that pharmaceutical companies are In 1934 Butenandt and Dannenbaum isolated not interested in supporting research in this field. dehydroepiandrosterone (DHEA) from urine and in 1944 2) DHEA can be described as a “human molecule” Munson and colleagues identified its 3β-sulphate because other investigated species have much lower (DHEAS). Even now, nearly 70 years later, we still do concentrations. -

COVID-19—The Potential Beneficial Therapeutic Effects of Spironolactone During SARS-Cov-2 Infection
pharmaceuticals Review COVID-19—The Potential Beneficial Therapeutic Effects of Spironolactone during SARS-CoV-2 Infection Katarzyna Kotfis 1,* , Kacper Lechowicz 1 , Sylwester Drozd˙ zal˙ 2 , Paulina Nied´zwiedzka-Rystwej 3 , Tomasz K. Wojdacz 4, Ewelina Grywalska 5 , Jowita Biernawska 6, Magda Wi´sniewska 7 and Miłosz Parczewski 8 1 Department of Anesthesiology, Intensive Therapy and Acute Intoxications, Pomeranian Medical University in Szczecin, 70-111 Szczecin, Poland; [email protected] 2 Department of Pharmacokinetics and Monitored Therapy, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] 3 Institute of Biology, University of Szczecin, 71-412 Szczecin, Poland; [email protected] 4 Independent Clinical Epigenetics Laboratory, Pomeranian Medical University, 71-252 Szczecin, Poland; [email protected] 5 Department of Clinical Immunology and Immunotherapy, Medical University of Lublin, 20-093 Lublin, Poland; [email protected] 6 Department of Anesthesiology and Intensive Therapy, Pomeranian Medical University in Szczecin, 71-252 Szczecin, Poland; [email protected] 7 Clinical Department of Nephrology, Transplantology and Internal Medicine, Pomeranian Medical University, 70-111 Szczecin, Poland; [email protected] 8 Department of Infectious, Tropical Diseases and Immune Deficiency, Pomeranian Medical University in Szczecin, 71-455 Szczecin, Poland; [email protected] * Correspondence: katarzyna.kotfi[email protected]; Tel.: +48-91-466-11-44 Abstract: In March 2020, coronavirus disease 2019 (COVID-19) caused by SARS-CoV-2 was declared Citation: Kotfis, K.; Lechowicz, K.; a global pandemic by the World Health Organization (WHO). The clinical course of the disease is Drozd˙ zal,˙ S.; Nied´zwiedzka-Rystwej, unpredictable but may lead to severe acute respiratory infection (SARI) and pneumonia leading to P.; Wojdacz, T.K.; Grywalska, E.; acute respiratory distress syndrome (ARDS). -

Effect of Dehydroepiandrosterone and Testosterone Supplementation on Systemic Lipolysis
ORIGINAL ARTICLE Effect of Dehydroepiandrosterone and Testosterone Supplementation on Systemic Lipolysis Ana E. Espinosa De Ycaza, Robert A. Rizza, K. Sreekumaran Nair, and Michael D. Jensen Division of Endocrinology, Endocrine Research Unit, Mayo Clinic, Rochester, Minnesota 55905 Downloaded from https://academic.oup.com/jcem/article/101/4/1719/2804555 by guest on 24 September 2021 Context: Dehydroepiandrosterone (DHEA) and T hormones are advertised as antiaging, antiobe- sity products. However, the evidence that these hormones have beneficial effects on adipose tissue metabolism is limited. Objective: The objective of the study was to determine the effect of DHEA and T supplementation on systemic lipolysis during a mixed-meal tolerance test (MMTT) and an iv glucose tolerance test (IVGTT). Design: This was a 2-year randomized, double-blind, placebo-controlled trial. Setting: The study was conducted at a general clinical research center. Participants: Sixty elderly women with low DHEA concentrations and 92 elderly men with low DHEA and bioavailable T concentrations participated in the study. Interventions: Elderly women received 50 mg DHEA (n ϭ 30) or placebo (n ϭ 30). Elderly men received 75 mg DHEA (n ϭ 30),5mgT(nϭ 30), or placebo (n ϭ 32). Main Outcome Measures: In vivo measures of systemic lipolysis (palmitate rate of appearance) during a MMTT or IVGTT. Results: At baseline there was no difference in insulin suppression of lipolysis measured during MMTT and IVGTT between the treatment groups and placebo. For both sexes, a univariate analysis showed no difference in changes in systemic lipolysis during the MMTT or IVGTT in the DHEA group and T group when compared with placebo. -

Reproductive DHEA-S
Reproductive DHEA-S Analyte Information - 1 - DHEA-S Introduction DHEA-S, DHEA sulfate or dehydroepiandrosterone sulfate, it is a metabolite of dehydroepiandrosterone (DHEA) resulting from the addition of a sulfate group. It is the sulfate form of aromatic C19 steroid with 10,13-dimethyl, 3-hydroxy group and 17-ketone. Its chemical name is 3β-hydroxy-5-androsten-17-one sulfate, its summary formula is C19H28O5S and its molecular weight (Mr) is 368.5 Da. The structural formula of DHEA-S is shown in (Fig.1). Fig.1: Structural formula of DHEA-S Other names used for DHEA-S include: Dehydroisoandrosterone sulfate, (3beta)-3- (sulfooxy), androst-5-en-17-one, 3beta-hydroxy-androst-5-en-17-one hydrogen sulfate, Prasterone sulfate and so on. As DHEA-S is very closely connected with DHEA, both hormones are mentioned together in the following text. Biosynthesis DHEA-S is the major C19 steroid and is a precursor in testosterone and estrogen biosynthesis. DHEA-S originates almost exclusively in the zona reticularis of the adrenal cortex (Fig.2). Some may be produced by the testes, none is produced by the ovaries. The adrenal gland is the sole source of this steroid in women, whereas in men the testes secrete 5% of DHEA-S and 10 – 20% of DHEA. The production of DHEA-S and DHEA is regulated by adrenocorticotropin (ACTH). Corticotropin-releasing hormone (CRH) and, to a lesser extent, arginine vasopressin (AVP) stimulate the release of adrenocorticotropin (ACTH) from the anterior pituitary gland (Fig.3). In turn, ACTH stimulates the adrenal cortex to secrete DHEA and DHEA-S, in addition to cortisol. -

Safety Data Sheet
SAFETY DATA SHEET SECTION 1: PRODUCT IDENTIFICATION PRODUCT NAME DHEA (Prasterone) (Micronized) PRODUCT CODE 0733 SUPPLIER MEDISCA Inc. Tel.: 1.800.932.1039 | Fax.: 1.855.850.5855 661 Route 3, Unit C, Plattsburgh, NY, 12901 3955 W. Mesa Vista Ave., Unit A-10, Las Vegas, NV, 89118 6641 N. Belt Line Road, Suite 130, Irving, TX, 75063 MEDISCA Pharmaceutique Inc. Tel.: 1.800.665.6334 | Fax.: 514.338.1693 4509 Rue Dobrin, St. Laurent, QC, H4R 2L8 21300 Gordon Way, Unit 153/158, Richmond, BC V6W 1M2 MEDISCA Australia PTY LTD Tel.: 1.300.786.392 | Fax.: 61.2.9700.9047 Unit 7, Heritage Business Park 5-9 Ricketty Street, Mascot, NSW 2020 EMERGENCY PHONE CHEMTREC Day or Night Within USA and Canada: 1-800-424-9300 NSW Poisons Information Centre: 131 126 USES Adjuvant; Androgen SECTION 2: HAZARDS IDENTIFICATION GHS CLASSIFICATION Toxic to Reproduction (Category 2) PICTOGRAM SIGNAL WORD Warning HAZARD STATEMENT(S) Reproductive effector, prohormone. Suspected of damaging fertility or the unborn child. May cause harm to breast-fed children. Causes serious eye irritation. Causes skin and respiratory irritation. Very toxic to aquatic life with long lasting effects. AUSTRALIA-ONLY HAZARDS Not Applicable. PRECAUTIONARY STATEMENT(S) Prevention Wash thoroughly after handling. Obtain special instructions before use. Do not handle until all safety precautions have been read and understood. Do not breathe dusts or mists. Do not eat, drink or smoke when using this product. Avoid contact during pregnancy/while nursing. Wear protective gloves, protective clothing, eye protection, face protection. Avoid release to the environment. Response IF ON SKIN (HAIR): Wash with plenty of water. -
Guidance for the Format and Content of the Protocol of Non-Interventional
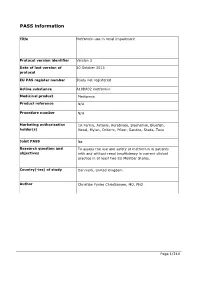
PASS information Title Metformin use in renal impairment Protocol version identifier Version 2 Date of last version of 30 October 2013 protocol EU PAS register number Study not registered Active substance A10BA02 metformin Medicinal product Metformin Product reference N/A Procedure number N/A Marketing authorisation 1A Farma, Actavis, Aurobindo, Biochemie, Bluefish, holder(s) Hexal, Mylan, Orifarm, Pfizer, Sandoz, Stada, Teva Joint PASS No Research question and To assess the use and safety of metformin in patients objectives with and without renal insufficiency in current clinical practice in at least two EU Member States. Country(-ies) of study Denmark, United Kingdom Author Christian Fynbo Christiansen, MD, PhD Page 1/214 Marketing authorisation holder(s) Marketing authorisation N/A holder(s) MAH contact person N/A Page 2/214 1. Table of Contents PASS information .......................................................................................................... 1 Marketing authorisation holder(s) .................................................................................... 2 1. Table of Contents ...................................................................................................... 3 2. List of abbreviations ................................................................................................... 4 3. Responsible parties .................................................................................................... 5 4. Abstract .................................................................................................................. -

A10 Anabolic Steroids Hardcore Info
CONTENTS GENERAL INFORMATION 3 Anabolic steroids – What are they? 4 How do they Work? – Aromatisation 5 More molecules – More problems 6 The side effects of anabolic steroids 7 Women and anabolic steroids 8 Injecting steroids 9 Abscesses – Needle Exchanges 10 Intramuscular injection 11 Injection sites 12 Oral steroids – Cycles – Stacking 13 Diet 14 Where do steroids come from? Spotting a counterfeit 15 Drug Information – Drug dosage STEROIDS 16 Anadrol – Andriol 17 Anavar – Deca-Durabolin 18 Dynabolon – Durabolin – Dianabol 19 Esiclene – Equipoise 20 Primobolan Depot – Proviron – Primobolan orals – Pronobol 21 Sustanon – Stromba, Strombaject – Testosterone Cypionate Testosterone Enanthate 22 Testosterone Propionate – Testosterone Suspension 23 Trenbolone Acetate – Winstrol OTHER DRUGS 24 Aldactone – Arimidex 25 Clenbuterol – Cytomel 26 Ephedrine Hydrochloride – GHB 27 Growth Hormone 28 Insulin 30 Insulin-Like Growth Factor-1 – Human Chorionic Gonadotrophin 31 Tamoxifen – Nubain – Recreational Drugs 32 Steroids and the Law 34 Glossary ANABOLIC STEROIDS People use anabolic steroids for various reasons, some use them to build muscle for their job, others just want to look good and some use them to help them in sport or body building. Whatever the reason, care needs to be taken so that as little harm is done to the body as possible because despite having muscle building effects they also have serious side effects especially when used incorrectly. WHAT ARE THEY? Anabolic steroids are man made versions of the hormone testosterone. Testosterone is the chemical in men responsible for facial hair, deepening of the voice and sex organ development, basically the masculine things Steroids are in a man. used in medicine to treat anaemia, muscle weakness after These masculine effects surgery etc, vascular are called the androgenic disorders and effects of testosterone. -

Dihydrotestosterone (Adractim®) 2.5% Gel for Topical Application
d g t squeezin l aa StSt gelgel Star gel from this en 0mg0m0mg 2.2.55 5mg5mg 7.7.55 10m1 mg l l 12.12.55 ge g ge ) 15m15mg f 17.17.55 ® 20m20mg g g o 22.22.55 25 25m25mg 1.25 g of g 1.25 1 1.25 1 1 1.25 g of g 1.25 27.5 30mg300mm 32.32.5.5 35m35mg drotestosterone hy 37.5 Put on a pair of gloves Gently squeeze the gel onto the mark on the ruler illustrated below – as instructed on the dispensing label. Leave for five minutes Wipe this laminated information sheet with a damp piece of kitchen paper ready for the next dose. Remove the gloves and wash them in warm soapy water ready for the next dose. of di 40m40mg © GOSH NHS Foundation Trust January 2016 © GOSH NHS Foundation Trust How is it used? Dihydrotestosterone 2.5% gel is for external It should be applied over the use only. required area of skin after washing. The gel should be left to dry for five minutes or so before putting on clothes. 1. 2. 3. Spread over the required area evenly 4. 5. Note: Dihydrotestosterone 2.5% gel should not be applied to any broken areas of skin. 42.42.55 l l 45mg45m45m ge ge 47.47.55 , ® 50mg50mm 52.52.55 55mg55mm 57.57.55 2.5 g of g 2 2.5 2 2.5 g of g 2.5 60mg600m0m 62.62.5.5 Dihydrotestosterone is a synthetic version of a hormone called testosterone. -

Spironolactone 2021 Newborn Use Only
Spironolactone 2021 Newborn use only Alert Spironolactone is a potassium-sparing diuretic and concomitant intake of potassium or ACE inhibitors may lead to hyperkalemia. Indication Diuretic primarily prescribed for its potassium-sparing effect. For heart failure, in conjunction with furosemide. For chronic lung disease, in conjunction with a thiazide diuretic. Bartter syndrome and Gitelman Syndrome. Action Spironolactone is a synthetic steroid that acts as a competitive aldosterone receptor antagonist, so inhibits sodium reabsorption and spares potassium. It is a weak diuretic. It also inhibits the interaction between dihydrotestosterone and the intracellular androgen receptor resulting in moderate antiandrogenic activity. Drug type Non-selective mineralocorticoid receptor antagonist. Trade name Aldactone; Spiractin Presentation Oral suspension prepared in pharmacy: 1 mg/mL; 2.5 mg/mL; 5 mg/mL. Dose 1–3 mg/kg/dose 24 hourly. Dose can be divided into different intervals. Dose adjustment Therapeutic hypothermia – Not applicable. Renal impairment – Refer to precautions section. Hepatic impairment – Refer to precautions section. Maximum dose 3 mg/kg/day Total cumulative dose Route Oral Preparation Oral suspension. Administration Administer undiluted with feeds. Monitoring Serum potassium at regular intervals. Contraindications Hyperkalaemia. Significant renal impairment. Anuria. Adrenal insufficiency. Precautions Use with caution in infants with renal or hepatic impairment. Monitor more frequently if infant is also given potassium. Drug interactions Spironolactone increases the effects of ACE inhibitors (leading to hyperkalemia), digoxin and sotalol. Adverse reactions Hyperkalaemia and metabolic acidosis. Antiandrogenic effects include reduced hirsutism and gynecomastia. There is one case report of an ovarian cyst in a neonate on spironolactone. Spironolactone interferes with 17-hydroxyprogesterone measurement, which is used to screen neonates for congenital adrenal hyperplasia. -

DHEA Sulfate, and Aging: Contribution of the Dheage Study to a Sociobiomedical Issue
Dehydroepiandrosterone (DHEA), DHEA sulfate, and aging: Contribution of the DHEAge Study to a sociobiomedical issue Etienne-Emile Baulieua,b, Guy Thomasc, Sylvie Legraind, Najiba Lahloue, Marc Rogere, Brigitte Debuiref, Veronique Faucounaug, Laurence Girardh, Marie-Pierre Hervyi, Florence Latourj, Marie-Ce´ line Leaudk, Amina Mokranel, He´ le` ne Pitti-Ferrandim, Christophe Trivallef, Olivier de Lacharrie` ren, Stephanie Nouveaun, Brigitte Rakoto-Arisono, Jean-Claude Souberbiellep, Jocelyne Raisonq, Yves Le Boucr, Agathe Raynaudr, Xavier Girerdq, and Franc¸oise Foretteg,j aInstitut National de la Sante´et de la Recherche Me´dicale Unit 488 and Colle`ge de France, 94276 Le Kremlin-Biceˆtre, France; cInstitut National de la Sante´et de la Recherche Me´dicale Unit 444, Hoˆpital Saint-Antoine, 75012 Paris, France; dHoˆpital Bichat, 75877 Paris, France; eHoˆpital Saint-Vincent de Paul, 75014 Paris, France; fHoˆpital Paul Brousse, 94804 Villejuif, France; gFondation Nationale de Ge´rontologie, 75016 Paris, France; hHoˆpital Charles Foix, 94205 Ivry, France; iHoˆpital de Biceˆtre, 94275 Biceˆtre, France; jHoˆpital Broca, 75013 Paris, France; kCentre Jack-Senet, 75015 Paris, France; lHoˆpital Sainte-Perine, 75016 Paris, France; mObservatoire de l’Age, 75017 Paris, France; nL’Ore´al, 92583 Clichy, France; oInstitut de Sexologie, 75116 Paris, France; pHoˆpital Necker, 75015 Paris, France; qHoˆpital Broussais, 75014 Paris, France; and rHoˆpital Trousseau, 75012 Paris, France Contributed by Etienne-Emile Baulieu, December 23, 1999 The secretion and the blood levels of the adrenal steroid dehydro- number of consumers. Extravagant publicity based on fantasy epiandrosterone (DHEA) and its sulfate ester (DHEAS) decrease pro- (‘‘fountain of youth,’’ ‘‘miracle pill’’) or pseudoscientific asser- foundly with age, and the question is posed whether administration tion (‘‘mother hormone,’’ ‘‘antidote for aging’’) has led to of the steroid to compensate for the decline counteracts defects unfounded radical assertions, from superactivity (‘‘keep young,’’ associated with aging. -

Treated with Steroids Derived from Dehydroepiandrosterone
Proc. Natl. Acad. Sci. USA Vol. 92, pp. 6617-6619, July 1995 Medical Sciences Ergosteroids: Induction of thermogenic enzymes in liver of rats treated with steroids derived from dehydroepiandrosterone HENRY LARDY, BRUCE PARTRIDGE, NANCY KNEER, AND YONG WEI Institute for Enzyme Research, University of Wisconsin, 1710 University Avenue, Madison, WI 53705 Contributed by Henry Lardy, April 24, 1995 ABSTRACT Dehydroepiandrosterone (DHEA), an inter- above normal concentrations (16); they become hirsute and mediate in the biosynthesis of testosterone and estrogens, exhibit other signs of masculinization. exerts several physiological effects not involving the sex It was demonstrated by Tagliaferro et al. (20) that feeding hormones. When fed to rats it induces the thermogenic DHEA to rats enhanced heat production and decreased enzymes mitochondrial sn-glycerol-3-phosphate dehydroge- efficiency of food utilization. We therefore measured its effect nase and cytosolic malic enzyme in their livers. Animals and on the production of liver mitochondrial sn-glycerol-3- humans, and their excised tissues, are known to hydroxylate phosphate dehydrogenase and cytosolic malic enzyme, two DHEA at several positions and to interconvert 7a-hydroxy- enzymes that had been demonstrated (21-24) to be induced by DHEA, 7,8-hydroxy-DHEA, 7-oxo-DHEA, and the correspond- a classic thermogenic agent, the thyroid hormone. Both en- ing derivatives of androst-5-enediol. We report here that these zymes were increased severalfold in livers of rats fed a diet 7-oxygenated derivatives are active inducers of these thermo- containing 0.2-0.5% DHEA for 6 days (25, 26). A possible genic enzymes in rats and that the 7-oxo derivatives are more mechanism by which these enzymes function to decrease active than the parent steroids. -

Selection of a Mineralocorticoid Receptor Antagonist for Patients with Hypertension Or Heart Failure
European Journal of Heart Failure (2014) 16, 143–150 REVIEW doi:10.1111/ejhf.31 Selection of a mineralocorticoid receptor antagonist for patients with hypertension or heart failure Javaid Iqbal1*, Yasir Parviz1, Bertram Pitt2, John Newell-Price3, Abdallah Al-Mohammad1, and Faiez Zannad4 1Department of Cardiovascular Science at the University of Sheffield and Cardiology Department at Sheffield Teaching Hospitals NHS Trust, Sheffield,K; U 2Cardiovascular Centre, University of Michigan, Ann Arbor, MI, USA; 3Department of Human Metabolism at the University of Sheffield and Endocrinology Department at Sheffield Teaching Hospitals NHS Trust, Sheffield, UK; and 4INSERM, Centre d’Investigation Clinique and Centre Hospitalier Universitaire, and the Department of Cardiology, Nancy University, Université de Lorraine, Nancy, France Received 8 April 2013; revised 15July2013; accepted 19July2013; online publish-ahead-of-print 14 December 2013 Clinical trials have demonstrated morbidity and mortality benefits of mineralocorticoid receptor antagonists (MRAs) in patients with heart failure. These studies have used either spironolactone or eplerenone as the MRA. It is generally believed that these two agents have the same effects, and the data from studies using one drug could be extrapolated for the other. National and international guidelines do not generally discriminate between spironolactone and eplerenone, but strongly recommend using an MRA for patients with heart failure due to LV systolic dysfunction and post-infarct LV systolic dysfunction. There are no major clinical trials directly comparing the efficacy of these two drugs. This article aims to compare the pharmacokinetics and pharmacodynamics of spironolactone and eplerenone, and to analyse the available data for their cardiovascular indications and adverse effects.